Department of Biomedical Informatics, 460 W 12th Avenue, 212 BRT, The Ohio State University, Columbus, OH 43210, USA.
Nucleic Acids Res. 2012 Sep;40(16):7690-704. doi: 10.1093/nar/gks501. Epub 2012 Jun 6.
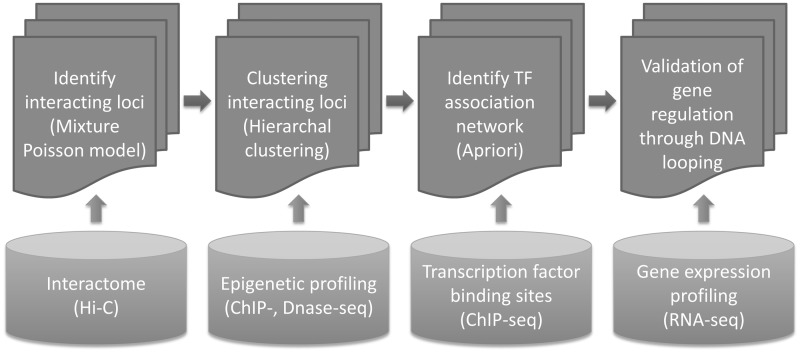
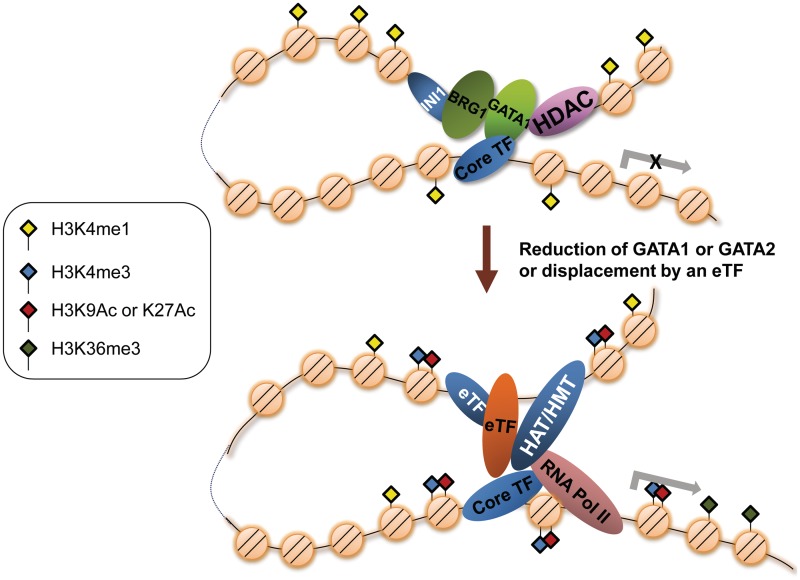
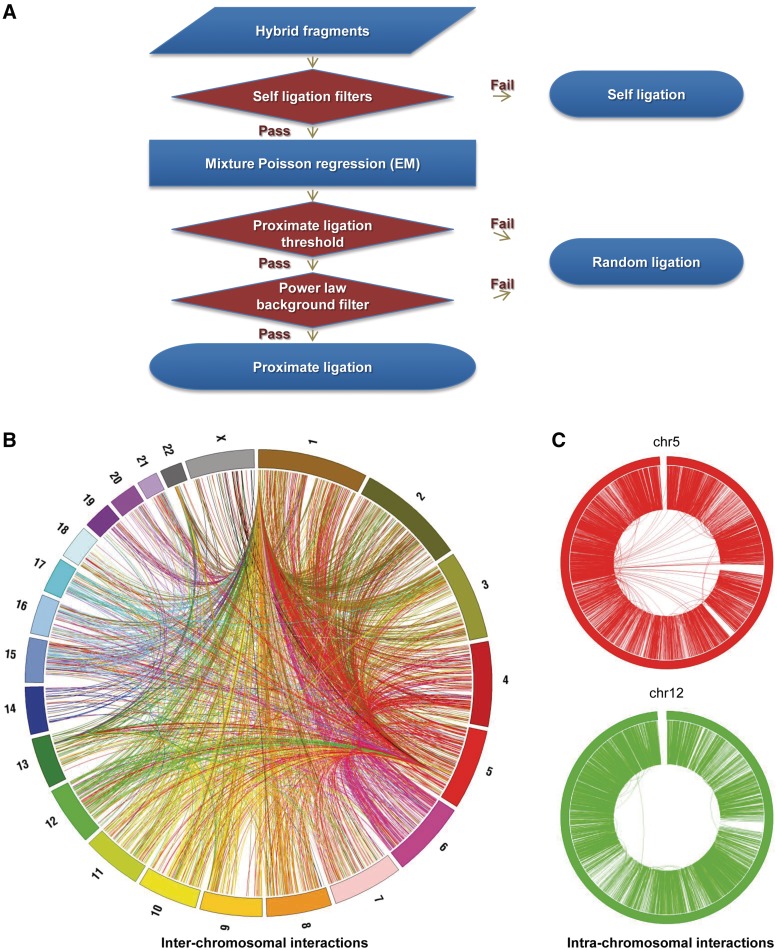
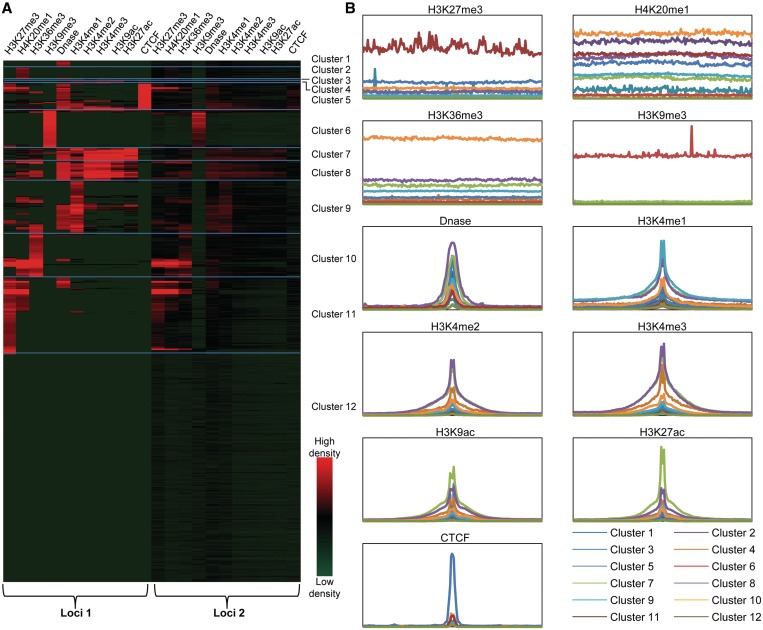
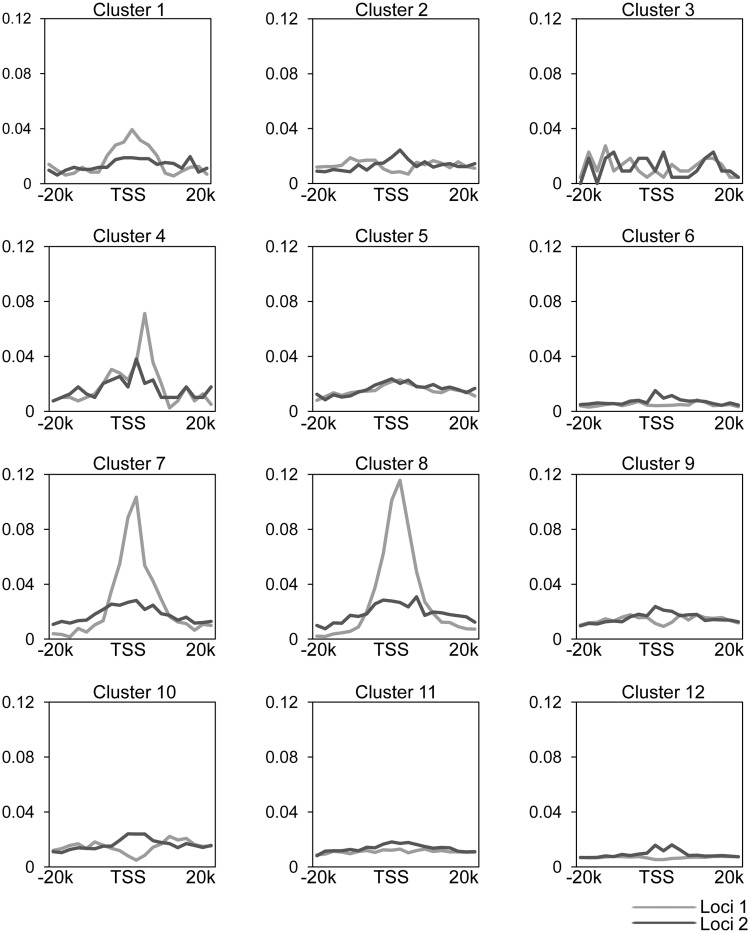
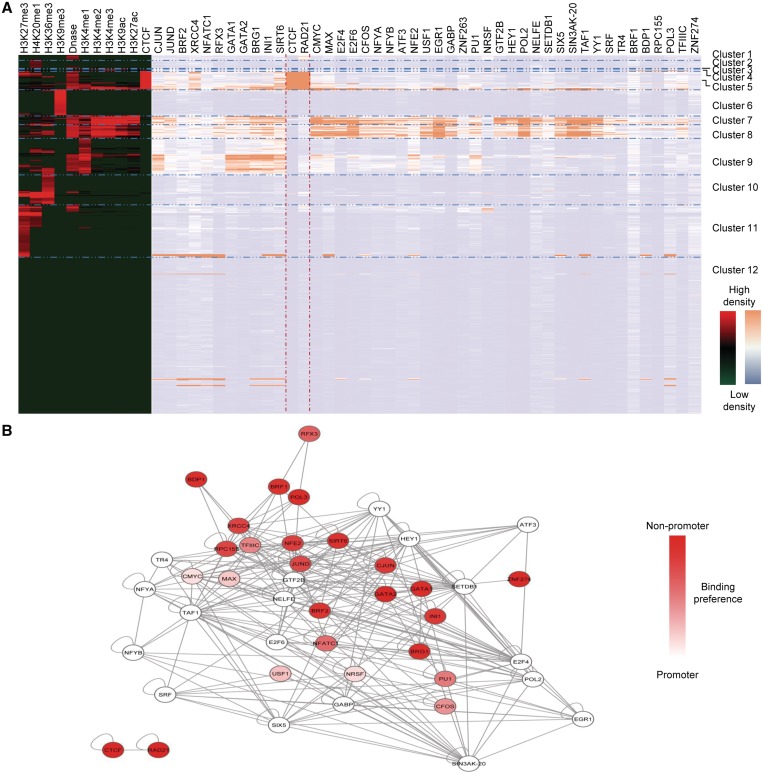
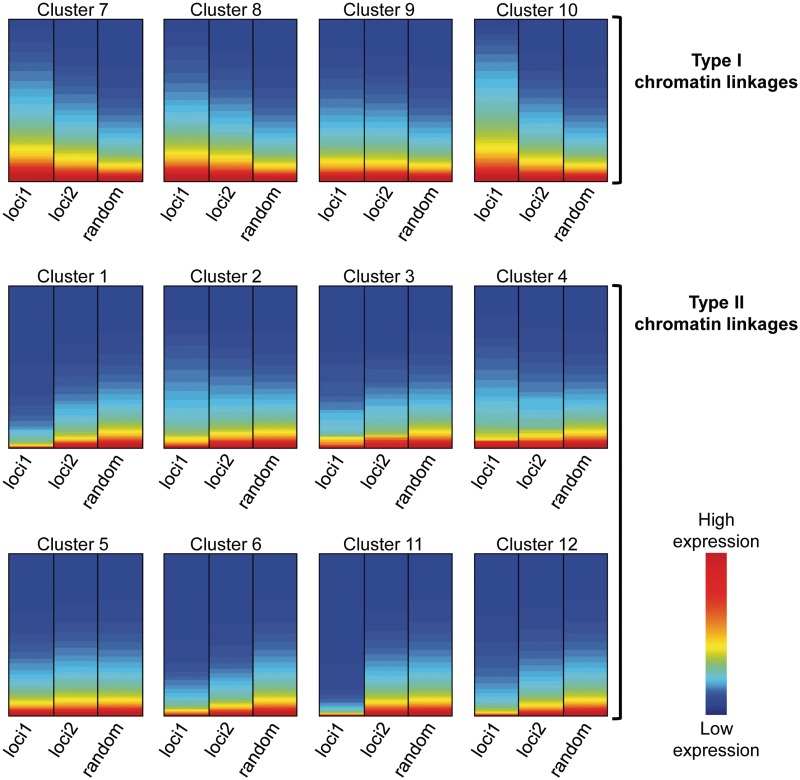
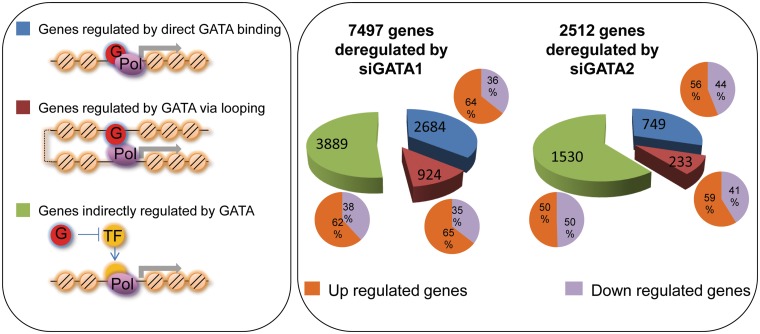
We have analyzed publicly available K562 Hi-C data, which enable genome-wide unbiased capturing of chromatin interactions, using a Mixture Poisson Regression Model and a power-law decay background to define a highly specific set of interacting genomic regions. We integrated multiple ENCODE Consortium resources with the Hi-C data, using DNase-seq data and ChIP-seq data for 45 transcription factors and 9 histone modifications. We classified 12 different sets (clusters) of interacting loci that can be distinguished by their chromatin modifications and which can be categorized into two types of chromatin linkages. The different clusters of loci display very different relationships with transcription factor-binding sites. As expected, many of the transcription factors show binding patterns specific to clusters composed of interacting loci that encompass promoters or enhancers. However, cluster 9, which is distinguished by marks of open chromatin but not by active enhancer or promoter marks, was not bound by most transcription factors but was highly enriched for three transcription factors (GATA1, GATA2 and c-Jun) and three chromatin modifiers (BRG1, INI1 and SIRT6). To investigate the impact of chromatin organization on gene regulation, we performed ribonucleicacid-seq analyses before and after knockdown of GATA1 or GATA2. We found that knockdown of the GATA factors not only alters the expression of genes having a nearby bound GATA but also affects expression of genes in interacting loci. Our work, in combination with previous studies linking regulation by GATA factors with c-Jun and BRG1, provides genome-wide evidence that Hi-C data identify sets of biologically relevant interacting loci.
我们分析了公开的 K562 Hi-C 数据,这些数据可使用混合泊松回归模型和幂律衰减背景来捕获全基因组的无偏染色质相互作用,从而定义高度特异性的相互作用基因组区域集。我们整合了多个 ENCODE 联盟资源与 Hi-C 数据,使用 DNase-seq 数据和 45 个转录因子和 9 个组蛋白修饰的 ChIP-seq 数据。我们将 12 个不同的相互作用基因座集(簇)进行分类,这些基因座集可以通过其染色质修饰来区分,并可以分为两种类型的染色质连接。不同的基因座簇与转录因子结合位点显示出非常不同的关系。正如预期的那样,许多转录因子显示出与包含启动子或增强子的相互作用基因座簇组成的特定结合模式。然而,cluster 9 与活跃的增强子或启动子标记不同,它的特征是开放染色质标记,但不受大多数转录因子的结合,而是高度富集了三个转录因子(GATA1、GATA2 和 c-Jun)和三个染色质修饰因子(BRG1、INI1 和 SIRT6)。为了研究染色质组织对基因调控的影响,我们在敲低 GATA1 或 GATA2 前后进行了核糖核酸-seq 分析。我们发现,敲低 GATA 因子不仅改变了附近结合 GATA 的基因的表达,还影响了相互作用基因座中基因的表达。我们的工作与以前将 GATA 因子的调节与 c-Jun 和 BRG1 联系起来的研究相结合,提供了全基因组证据,表明 Hi-C 数据可以识别具有生物学相关性的相互作用基因座集。