Department of Cancer Research and Molecular Medicine, Norwegian University of Science and Technology, Trondheim, Norway.
BMC Biol. 2011 Nov 24;9:80. doi: 10.1186/1741-7007-9-80.
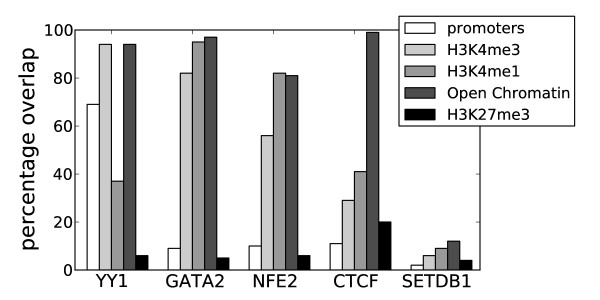
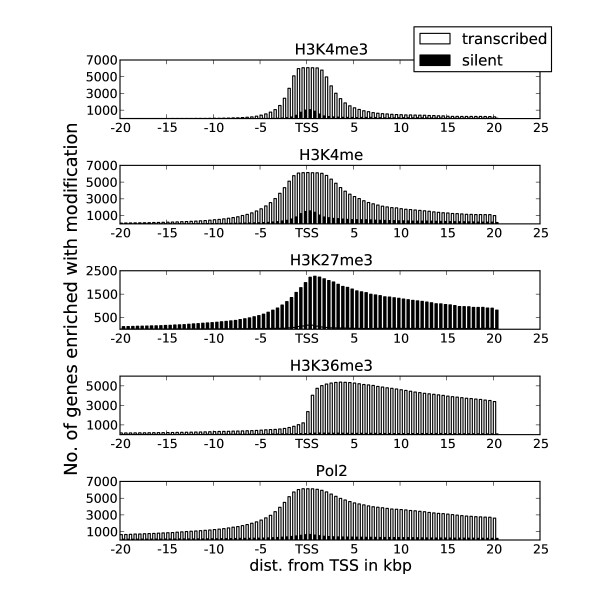
Transcription factor binding to DNA requires both an appropriate binding element and suitably open chromatin, which together help to define regulatory elements within the genome. Current methods of identifying regulatory elements, such as promoters or enhancers, typically rely on sequence conservation, existing gene annotations or specific marks, such as histone modifications and p300 binding methods, each of which has its own biases.
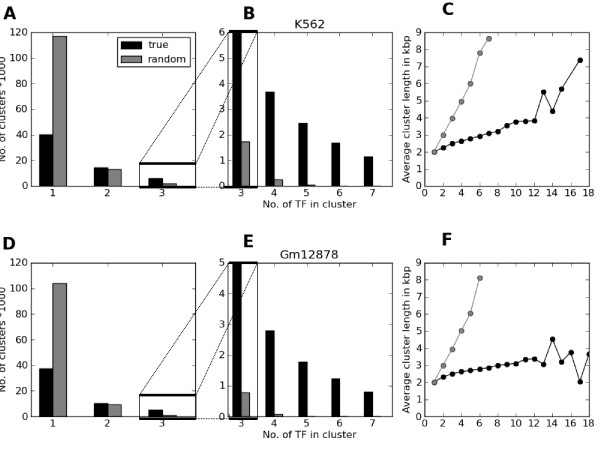
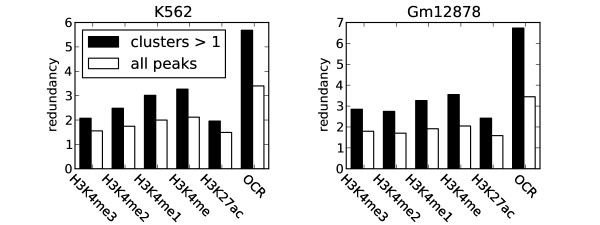
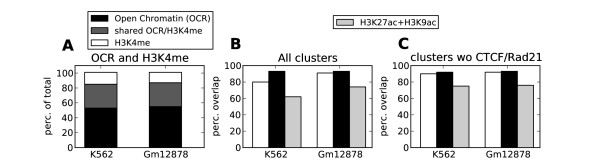
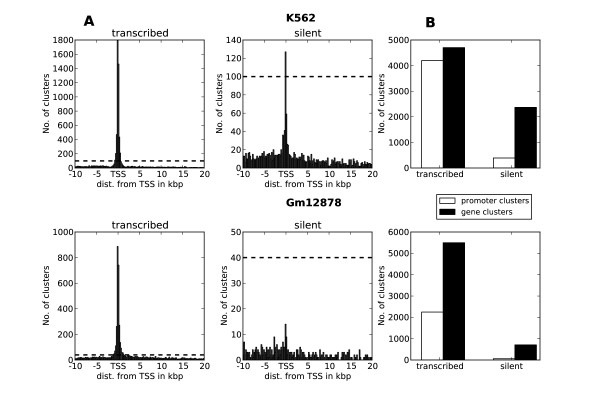
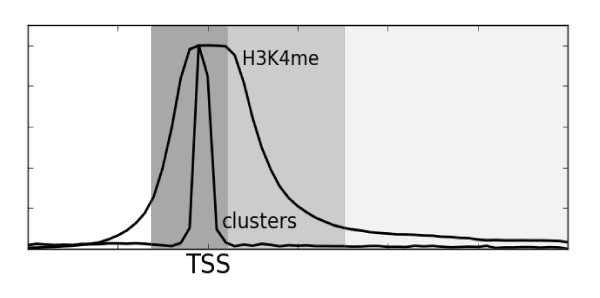
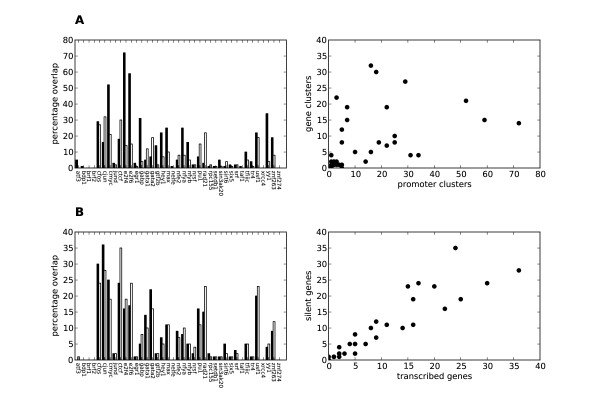
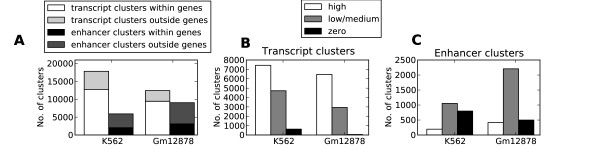
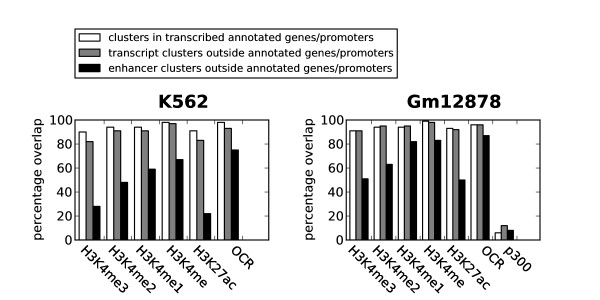
Herein we show that an approach based on clustering of transcription factor peaks from high-throughput sequencing coupled with chromatin immunoprecipitation (Chip-Seq) can be used to evaluate markers for regulatory elements. We used 67 data sets for 54 unique transcription factors distributed over two cell lines to create regulatory element clusters. By integrating the clusters from our approach with histone modifications and data for open chromatin, we identified general methylation of lysine 4 on histone H3 (H3K4me) as the most specific marker for transcription factor clusters. Clusters mapping to annotated genes showed distinct patterns in cluster composition related to gene expression and histone modifications. Clusters mapping to intergenic regions fall into two groups either directly involved in transcription, including miRNAs and long noncoding RNAs, or facilitating transcription by long-range interactions. The latter clusters were specifically enriched with H3K4me1, but less with acetylation of lysine 27 on histone 3 or p300 binding.
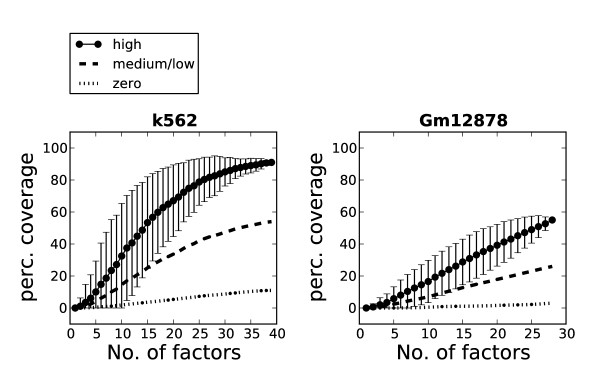
By integrating genomewide data of transcription factor binding and chromatin structure and using our data-driven approach, we pinpointed the chromatin marks that best explain transcription factor association with different regulatory elements. Our results also indicate that a modest selection of transcription factors may be sufficient to map most regulatory elements in the human genome.
转录因子与 DNA 的结合需要适当的结合元件和适当开放的染色质,这两者共同有助于在基因组内定义调控元件。目前识别调控元件(如启动子或增强子)的方法通常依赖于序列保守性、现有的基因注释或特定的标记,如组蛋白修饰和 p300 结合方法,每种方法都有其自身的偏向性。
本文展示了一种基于高通量测序中转录因子峰聚类与染色质免疫沉淀(Chip-Seq)相结合的方法,可用于评估调控元件的标记物。我们使用了两个细胞系中的 54 个独特转录因子的 67 个数据集来创建调控元件簇。通过整合我们的方法与组蛋白修饰和开放染色质的数据,我们发现组蛋白 H3 赖氨酸 4 上的整体甲基化(H3K4me)是转录因子簇最特异的标记物。注释基因的簇在与基因表达和组蛋白修饰相关的簇组成上表现出不同的模式。映射到基因间区域的簇分为两类,一类直接参与转录,包括 miRNA 和长非编码 RNA,另一类通过长距离相互作用促进转录。后者簇特异性地富集 H3K4me1,但乙酰化的赖氨酸 27 上的组蛋白 3 或 p300 结合较少。
通过整合转录因子结合和染色质结构的全基因组数据,并使用我们的数据驱动方法,我们确定了最能解释不同调控元件与转录因子关联的染色质标记物。我们的结果还表明,适度选择转录因子可能足以绘制人类基因组中的大多数调控元件。