Tello Javier A, Newton Claire L, Bouligand Jerome, Guiochon-Mantel Anne, Millar Robert P, Young Jacques
Centre for Integrative Physiology, School of Biomedical Sciences, University of Edinburgh, Edinburgh, United Kingdom.
PLoS One. 2012;7(6):e38456. doi: 10.1371/journal.pone.0038456. Epub 2012 Jun 5.
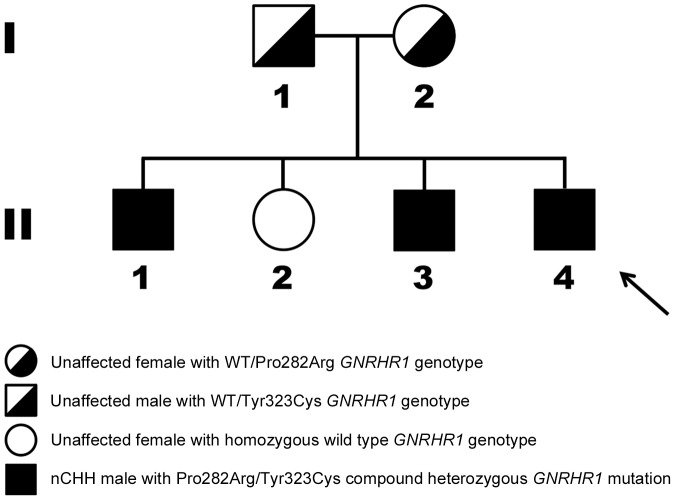
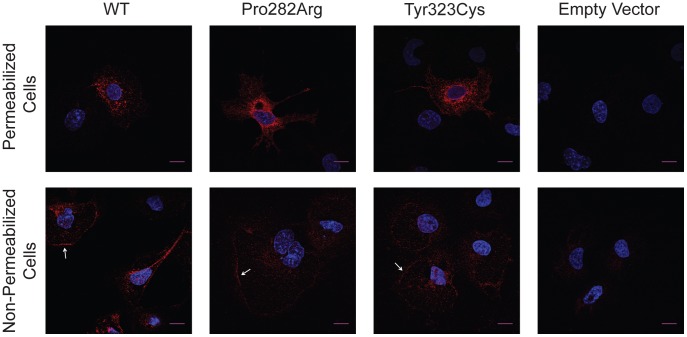
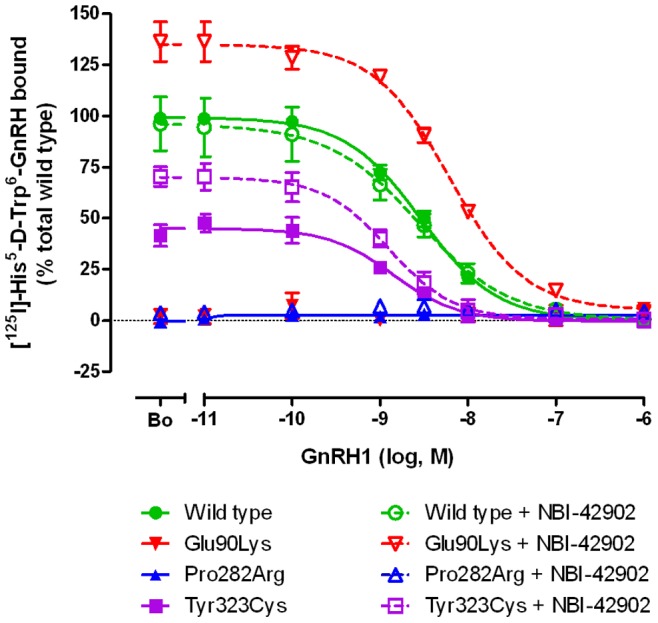
Congenital hypogonadotropic hypogonadism (CHH) is characterized by low gonadotropins and failure to progress normally through puberty. Mutations in the gene encoding the GnRH receptor (GNRHR1) result in CHH when present as compound heterozygous or homozygous inactivating mutations. This study identifies and characterizes the properties of two novel GNRHR1 mutations in a family in which three brothers display normosmic CHH while their sister was unaffected. Molecular analysis in the proband and the affected brothers revealed two novel non-synonymous missense GNRHR1 mutations, present in a compound heterozygous state, whereas their unaffected parents possessed only one inactivating mutation, demonstrating the autosomal recessive transmission in this kindred and excluding X-linked inheritance equivocally suggested by the initial pedigree analysis. The first mutation at c.845 C>G introduces an Arg substitution for the conserved Pro 282 in transmembrane domain (TMD) 6. The Pro282Arg mutant is unable to bind radiolabeled GnRH analogue. As this conserved residue is important in receptor conformation, it is likely that the mutation perturbs the binding pocket and affects trafficking to the cell surface. The second mutation at c.968 A>G introduces a Cys substitution for Tyr 323 in the functionally crucial N/DPxxY motif in TMD 7. The Tyr323Cys mutant has an increased GnRH binding affinity but reduced receptor expression at the plasma membrane and impaired G protein-coupling. Inositol phosphate accumulation assays demonstrated absent and impaired Gα(q/11) signal transduction by Pro282Arg and Tyr323Cys mutants, respectively. Pretreatment with the membrane permeant GnRHR antagonist NBI-42902, which rescues cell surface expression of many GNRHR1 mutants, significantly increased the levels of radioligand binding and intracellular signaling of the Tyr323Cys mutant but not Pro282Arg. Immunocytochemistry confirmed that both mutants are present on the cell membrane albeit at low levels. Together these molecular deficiencies of the two novel GNRHR1 mutations lead to the CHH phenotype when present as a compound heterozygote.
先天性低促性腺激素性性腺功能减退(CHH)的特征是促性腺激素水平低且青春期发育不能正常进行。编码促性腺激素释放激素受体(GNRHR1)的基因突变以复合杂合或纯合失活突变形式存在时会导致CHH。本研究鉴定并表征了一个家族中两个新的GNRHR1突变的特性,该家族中有三兄弟表现为嗅觉正常的CHH,而他们的妹妹未受影响。对先证者和患病兄弟的分子分析揭示了两个新的非同义错义GNRHR1突变,呈复合杂合状态,而他们未受影响的父母仅携带一个失活突变,表明该家族为常染色体隐性遗传,并排除了最初系谱分析中含糊暗示的X连锁遗传。第一个突变是c.845 C>G,导致跨膜结构域(TMD)6中保守的Pro 282被Arg取代。Pro282Arg突变体无法结合放射性标记的促性腺激素释放激素类似物。由于这个保守残基在受体构象中很重要,该突变很可能扰乱了结合口袋并影响了向细胞表面的转运。第二个突变是c.968 A>G,导致TMD 7中功能关键的N/DPxxY基序中的Tyr 323被Cys取代。Tyr323Cys突变体具有增加的促性腺激素释放激素结合亲和力,但质膜上的受体表达减少且G蛋白偶联受损。肌醇磷酸积累试验表明,Pro282Arg和Tyr323Cys突变体分别不存在和受损Gα(q/11)信号转导。用膜渗透性促性腺激素释放激素受体拮抗剂NBI-42902预处理可挽救许多GNRHR1突变体的细胞表面表达,显著增加了Tyr323Cys突变体而非Pro282Arg的放射性配体结合水平和细胞内信号传导。免疫细胞化学证实,这两个突变体均存在于细胞膜上,尽管水平较低。当这两个新的GNRHR1突变以复合杂合子形式存在时,这些分子缺陷共同导致了CHH表型。