Ludwig Institute for Cancer Research, Melbourne - Parkville Branch, Parkville, Victoria, Australia.
EMBO Mol Med. 2012 Sep;4(9):939-51. doi: 10.1002/emmm.201100604. Epub 2012 Jun 8.
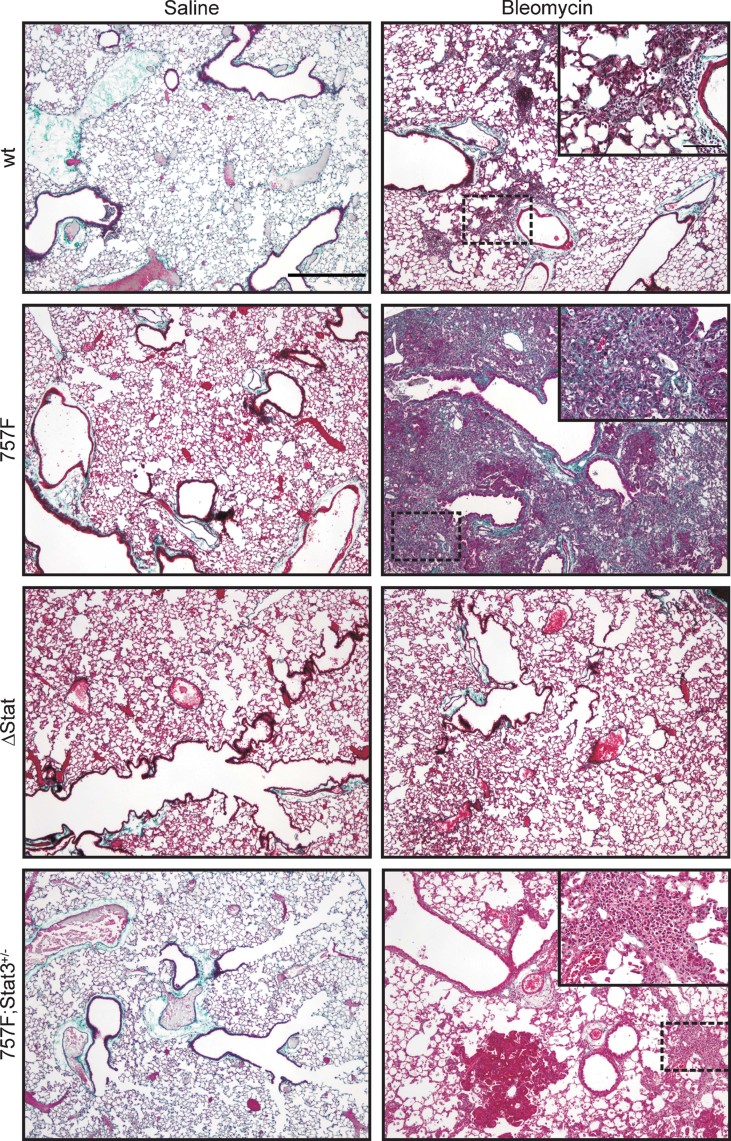
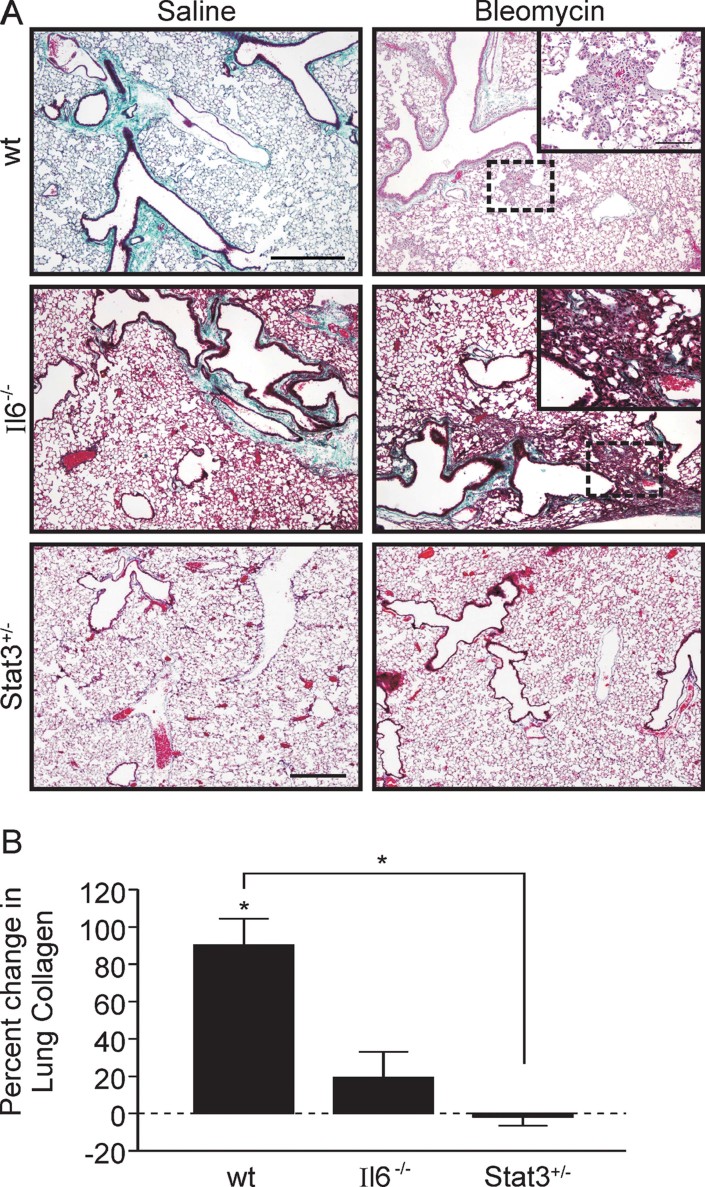
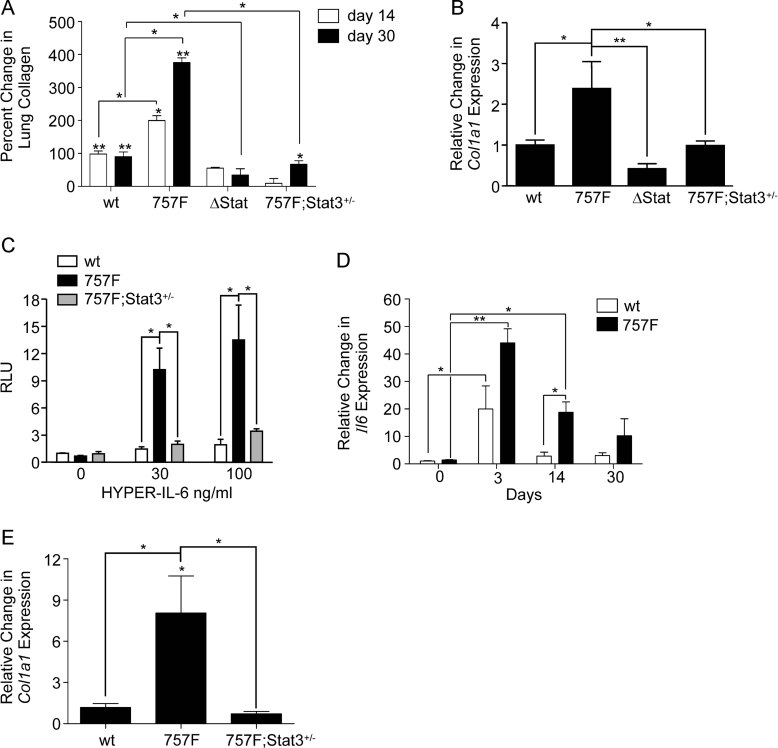
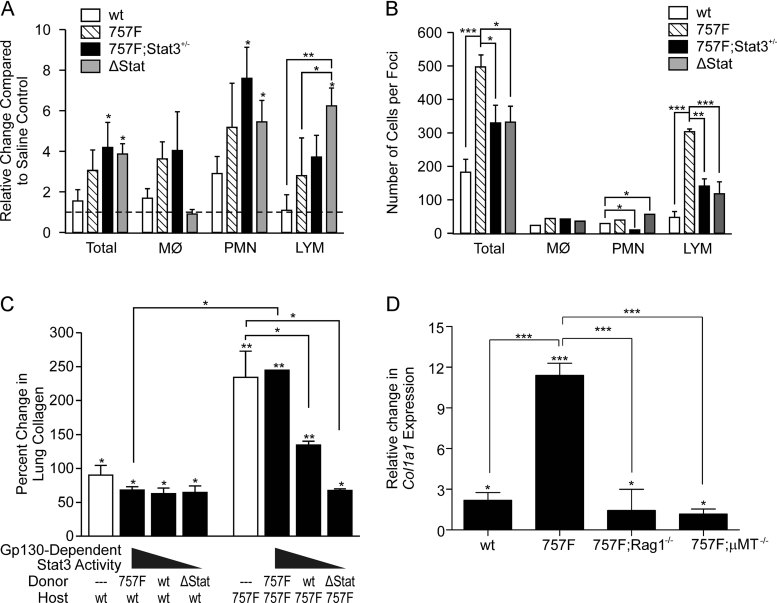
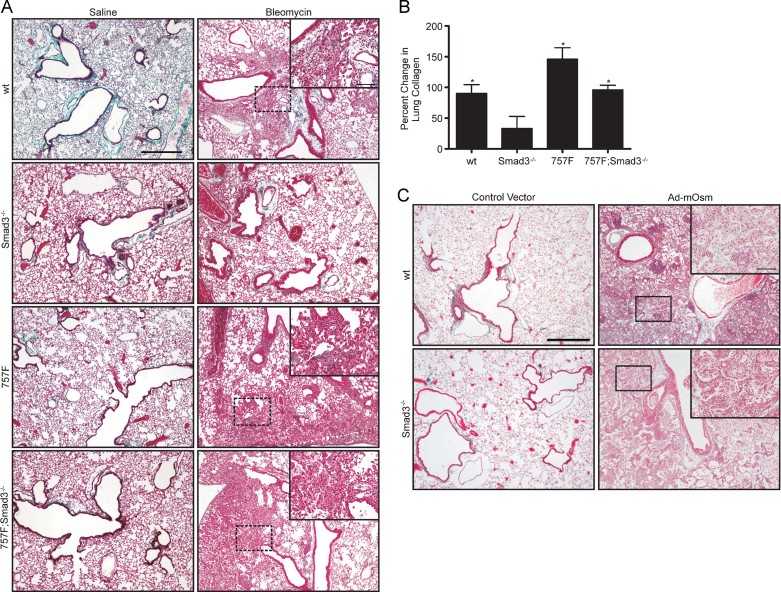
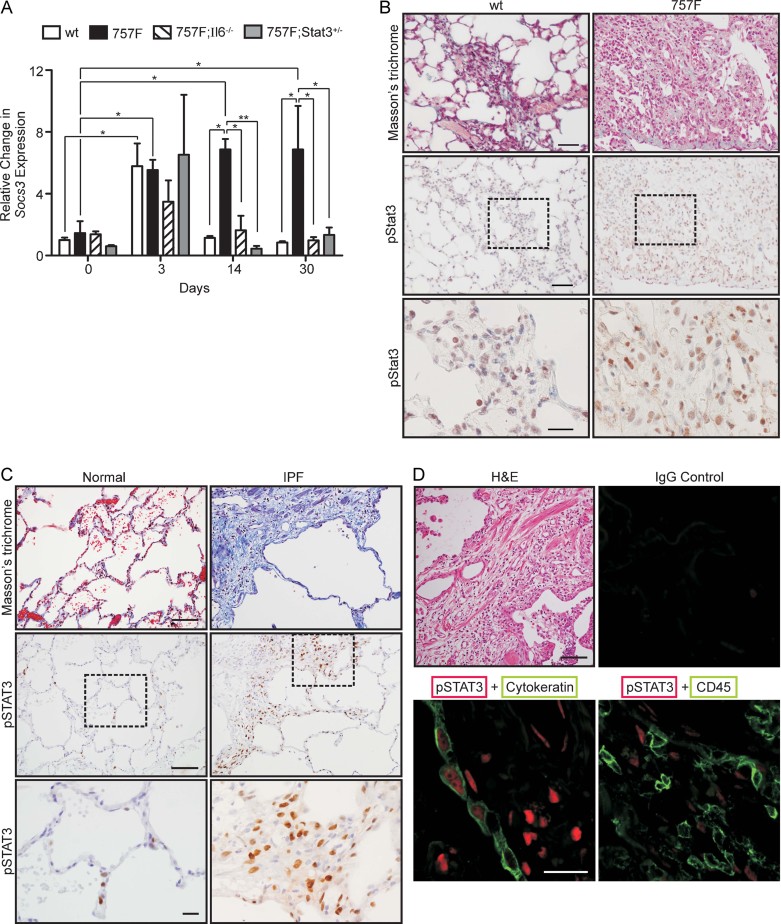
Idiopathic pulmonary fibrosis (IPF) is a fatal disease that is unresponsive to current therapies and characterized by excessive collagen deposition and subsequent fibrosis. While inflammatory cytokines, including interleukin (IL)-6, are elevated in IPF, the molecular mechanisms that underlie this disease are incompletely understood, although the development of fibrosis is believed to depend on canonical transforming growth factor (TGF)-β signalling. We examined bleomycin-induced inflammation and fibrosis in mice carrying a mutation in the shared IL-6 family receptor gp130. Using genetic complementation, we directly correlate the extent of IL-6-mediated, excessive Stat3 activity with inflammatory infiltrates in the lung and the severity of fibrosis in corresponding gp130(757F) mice. The extent of fibrosis was attenuated in B lymphocyte-deficient gp130(757F);µMT(-/-) compound mutant mice, but fibrosis still occurred in their Smad3(-/-) counterparts consistent with the capacity of excessive Stat3 activity to induce collagen 1α1 gene transcription independently of canonical TGF-β/Smad3 signalling. These findings are of therapeutic relevance, since we confirmed abundant STAT3 activation in fibrotic lungs from IPF patients and showed that genetic reduction of Stat3 protected mice from bleomycin-induced lung fibrosis.
特发性肺纤维化(IPF)是一种致命的疾病,对目前的治疗方法没有反应,其特征是胶原过度沉积和随后的纤维化。虽然白细胞介素(IL)-6 等炎症细胞因子在 IPF 中升高,但这种疾病的分子机制尚不完全清楚,尽管纤维化的发展被认为依赖于经典的转化生长因子(TGF)-β信号。我们研究了携带 IL-6 家族受体 gp130 突变的小鼠中的博莱霉素诱导的炎症和纤维化。通过遗传互补,我们直接将 IL-6 介导的、过度 Stat3 活性的程度与肺部的炎症浸润以及相应的 gp130(757F)小鼠的纤维化严重程度相关联。在 B 淋巴细胞缺陷型 gp130(757F);µMT(-/-)复合突变小鼠中,纤维化程度减轻,但在其 Smad3(-/-)对应物中仍发生纤维化,这与过度 Stat3 活性能够独立于经典 TGF-β/Smad3 信号诱导胶原 1α1 基因转录的能力一致。这些发现具有治疗相关性,因为我们证实了 IPF 患者纤维化肺中存在丰富的 STAT3 激活,并表明 Stat3 的遗传减少可保护小鼠免受博莱霉素诱导的肺纤维化。