Department of Computer Science, University of California, Los Angeles, CA 90095, USA.
Bioinformatics. 2012 Jun 15;28(12):i147-53. doi: 10.1093/bioinformatics/bts235.
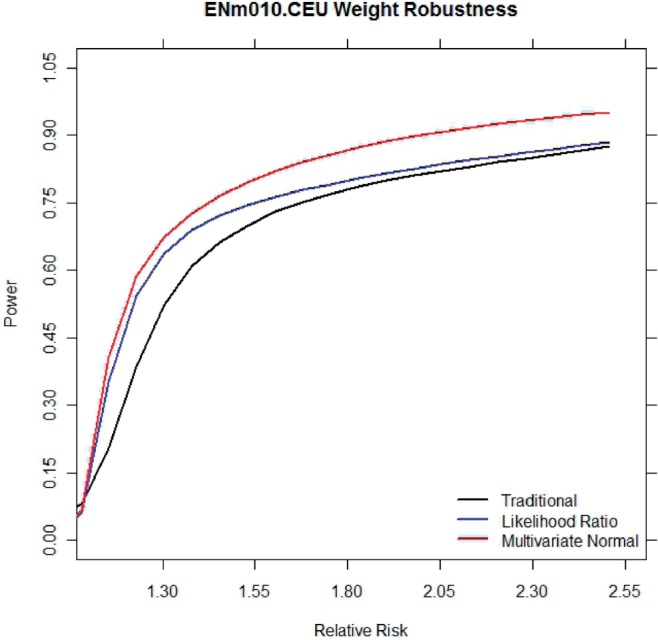
Recent technological developments in measuring genetic variation have ushered in an era of genome-wide association studies which have discovered many genes involved in human disease. Current methods to perform association studies collect genetic information and compare the frequency of variants in individuals with and without the disease. Standard approaches do not take into account any information on whether or not a given variant is likely to have an effect on the disease. We propose a novel method for computing an association statistic which takes into account prior information. Our method improves both power and resolution by 8% and 27%, respectively, over traditional methods for performing association studies when applied to simulations using the HapMap data. Advantages of our method are that it is as simple to apply to association studies as standard methods, the results of the method are interpretable as the method reports p-values, and the method is optimal in its use of prior information in regards to statistical power.
The method presented herein is available at http://masa.cs.ucla.edu.
最近在测量遗传变异方面的技术发展迎来了全基因组关联研究的时代,该研究发现了许多与人类疾病相关的基因。目前进行关联研究的方法收集遗传信息,并比较患病个体和无病个体中变异的频率。标准方法没有考虑到给定变异是否可能对疾病产生影响的任何信息。我们提出了一种计算关联统计量的新方法,该方法考虑了先验信息。当应用于基于 HapMap 数据的模拟时,与传统的关联研究方法相比,我们的方法分别将功效和分辨率提高了 8%和 27%。我们的方法的优点是,它与标准方法一样易于应用于关联研究,方法的结果可以解释为报告 p 值,并且该方法在使用先验信息方面在统计功效方面是最优的。
本文提出的方法可在 http://masa.cs.ucla.edu 上获得。