Department of Genetics, Washington University School of Medicine, St. Louis, MO 63108, USA.
Bioinformatics. 2012 Jun 15;28(12):i84-9. doi: 10.1093/bioinformatics/bts202.
Recognition models for protein-DNA interactions, which allow the prediction of specificity for a DNA-binding domain based only on its sequence or the alteration of specificity through rational design, have long been a goal of computational biology. There has been some progress in constructing useful models, especially for C(2)H(2) zinc finger proteins, but it remains a challenging problem with ample room for improvement. For most families of transcription factors the best available methods utilize k-nearest neighbor (KNN) algorithms to make specificity predictions based on the average of the specificities of the k most similar proteins with defined specificities. Homeodomain (HD) proteins are the second most abundant family of transcription factors, after zinc fingers, in most metazoan genomes, and as a consequence an effective recognition model for this family would facilitate predictive models of many transcriptional regulatory networks within these genomes.
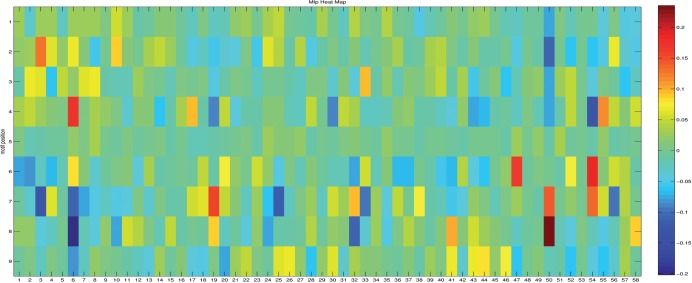
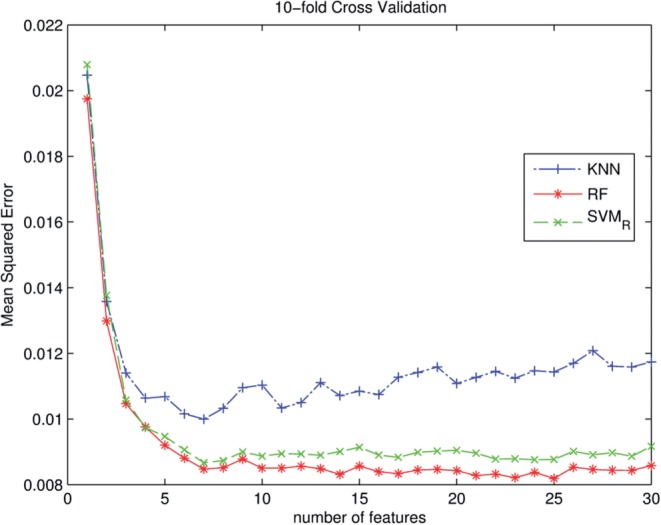
Using extensive experimental data, we have tested several machine learning approaches and find that both support vector machines and random forests (RFs) can produce recognition models for HD proteins that are significant improvements over KNN-based methods. Cross-validation analyses show that the resulting models are capable of predicting specificities with high accuracy. We have produced a web-based prediction tool, PreMoTF (Predicted Motifs for Transcription Factors) (http://stormo.wustl.edu/PreMoTF), for predicting position frequency matrices from protein sequence using a RF-based model.
识别蛋白质-DNA 相互作用的模型,仅基于其序列或通过合理设计改变特异性,允许预测 DNA 结合域的特异性,长期以来一直是计算生物学的目标。在构建有用的模型方面已经取得了一些进展,特别是对于 C(2)H(2)锌指蛋白,但这仍然是一个具有很大改进空间的具有挑战性的问题。对于大多数转录因子家族,最好的可用方法是使用 K-最近邻 (KNN) 算法根据具有明确定义特异性的 k 个最相似蛋白质的特异性平均值来进行特异性预测。同源域 (HD) 蛋白是大多数后生动物基因组中仅次于锌指的第二大转录因子家族,因此,针对该家族的有效识别模型将有助于预测这些基因组中许多转录调控网络的模型。
我们使用广泛的实验数据测试了几种机器学习方法,发现支持向量机和随机森林 (RF) 都可以为 HD 蛋白生成识别模型,这些模型比基于 KNN 的方法有显著改进。交叉验证分析表明,所得模型能够以高精度预测特异性。我们已经开发了一个基于网络的预测工具 PreMoTF(转录因子的预测基序)(http://stormo.wustl.edu/PreMoTF),用于使用基于 RF 的模型从蛋白质序列预测位置频率矩阵。