Dipartimento di Fisica, Sapienza Università di Roma, Roma, Italy.
PLoS Comput Biol. 2012;8(6):e1002562. doi: 10.1371/journal.pcbi.1002562. Epub 2012 Jun 21.
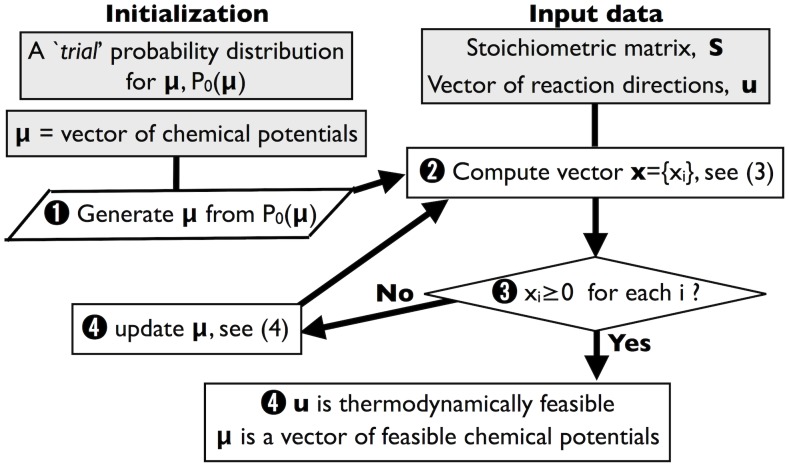
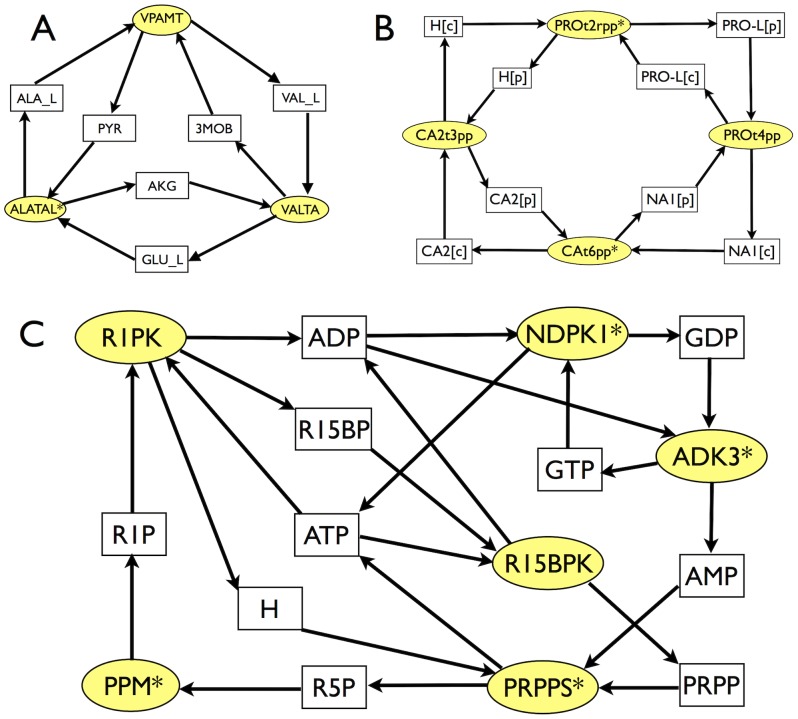
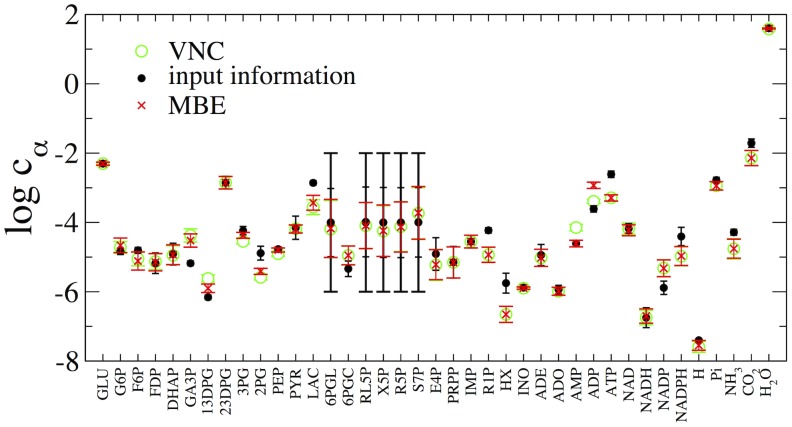
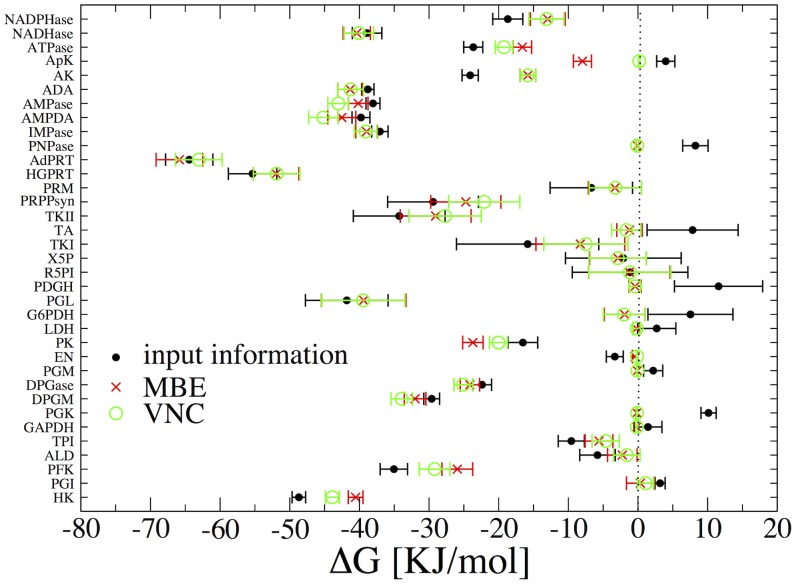
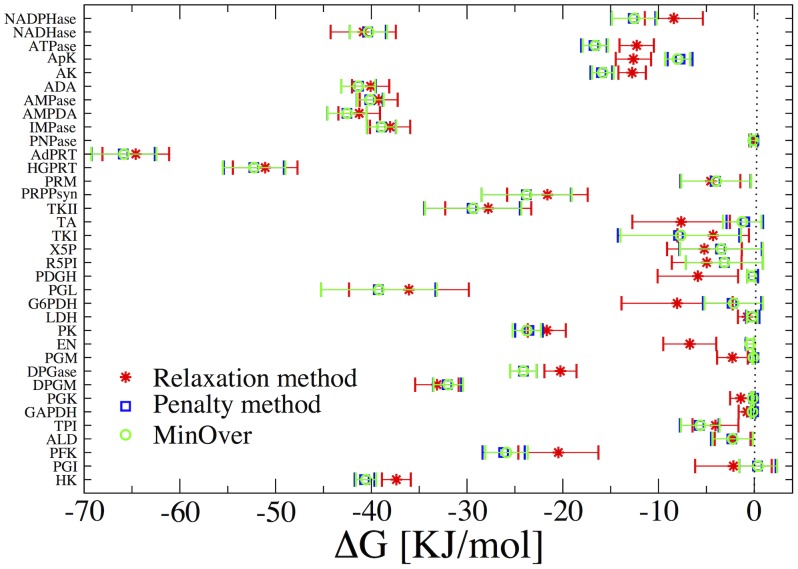
The integration of various types of genomic data into predictive models of biological networks is one of the main challenges currently faced by computational biology. Constraint-based models in particular play a key role in the attempt to obtain a quantitative understanding of cellular metabolism at genome scale. In essence, their goal is to frame the metabolic capabilities of an organism based on minimal assumptions that describe the steady states of the underlying reaction network via suitable stoichiometric constraints, specifically mass balance and energy balance (i.e. thermodynamic feasibility). The implementation of these requirements to generate viable configurations of reaction fluxes and/or to test given flux profiles for thermodynamic feasibility can however prove to be computationally intensive. We propose here a fast and scalable stoichiometry-based method to explore the Gibbs energy landscape of a biochemical network at steady state. The method is applied to the problem of reconstructing the Gibbs energy landscape underlying metabolic activity in the human red blood cell, and to that of identifying and removing thermodynamically infeasible reaction cycles in the Escherichia coli metabolic network (iAF1260). In the former case, we produce consistent predictions for chemical potentials (or log-concentrations) of intracellular metabolites; in the latter, we identify a restricted set of loops (23 in total) in the periplasmic and cytoplasmic core as the origin of thermodynamic infeasibility in a large sample (10(6)) of flux configurations generated randomly and compatibly with the prior information available on reaction reversibility.
将各种类型的基因组数据整合到生物网络的预测模型中是计算生物学目前面临的主要挑战之一。基于约束的模型在试图从定量上理解细胞代谢的基因组尺度方面尤其起着关键作用。从本质上讲,它们的目标是根据最小的假设来构建生物体的代谢能力,这些假设通过合适的化学计量约束(即质量平衡和能量平衡,也就是热力学可行性)来描述基础反应网络的稳态。然而,为了生成可行的反应通量配置或测试给定通量分布的热力学可行性,实现这些要求可能会非常耗费计算资源。我们在这里提出了一种快速且可扩展的基于化学计量的方法,用于探索生化网络在稳态下的吉布斯能量景观。该方法应用于重建人类红细胞代谢活性下的吉布斯能量景观的问题,以及识别和去除大肠杆菌代谢网络(iAF1260)中热力学不可行的反应循环的问题。在前一种情况下,我们对细胞内代谢物的化学势(或对数浓度)做出了一致的预测;在后一种情况下,我们在随机生成的、与可用的反应可逆性相关的先验信息兼容的大样本(10^6)通量配置中,确定了一个受限的环(总共 23 个),作为热力学不可行性的起源,这些环位于周质和细胞质核心中。