Department of Molecular Neuroscience, UCL Institute of Neurology, Queen Square, London WC1N 3BG, UK.
Cell Death Dis. 2012 Jun 28;3(6):e335. doi: 10.1038/cddis.2012.77.
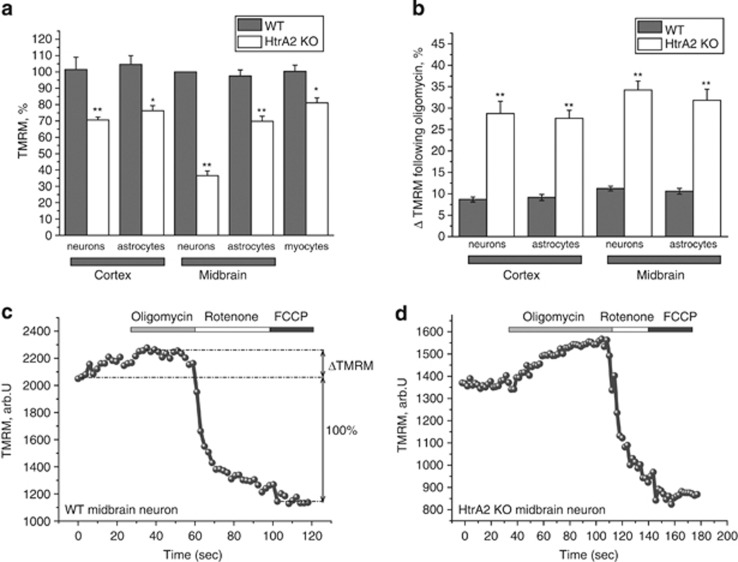
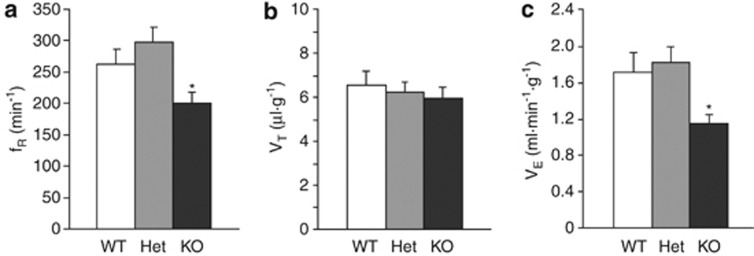
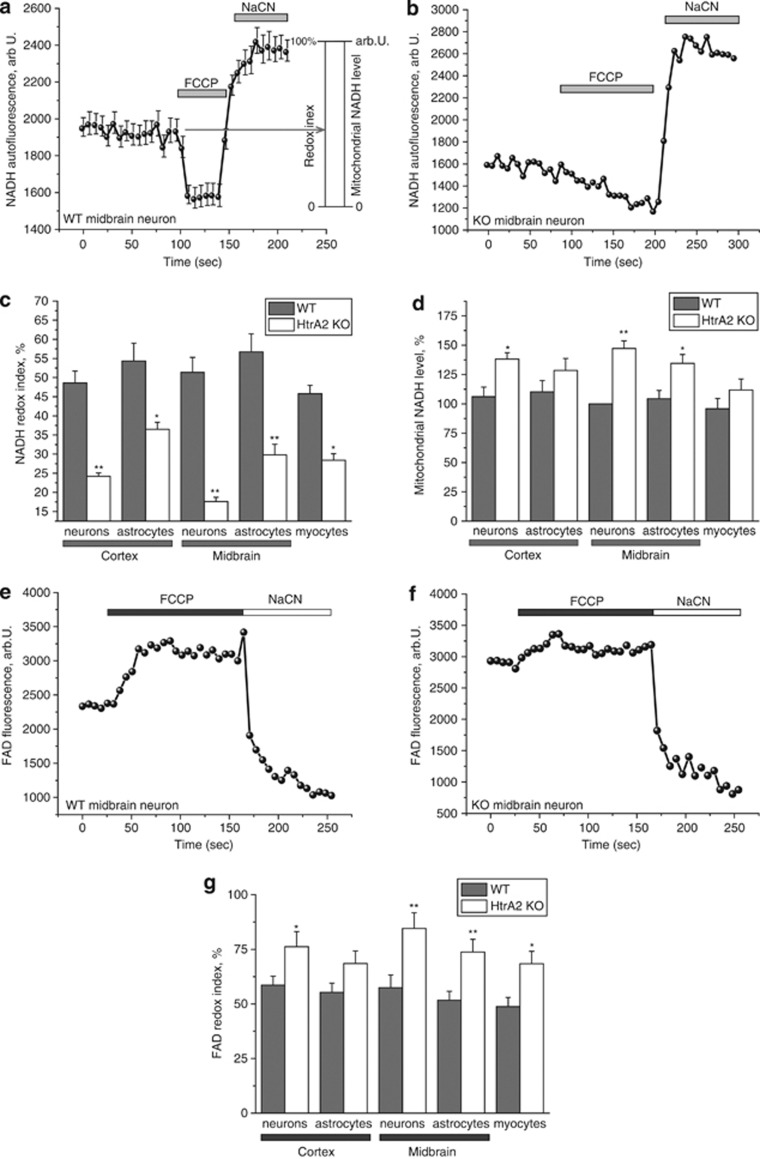
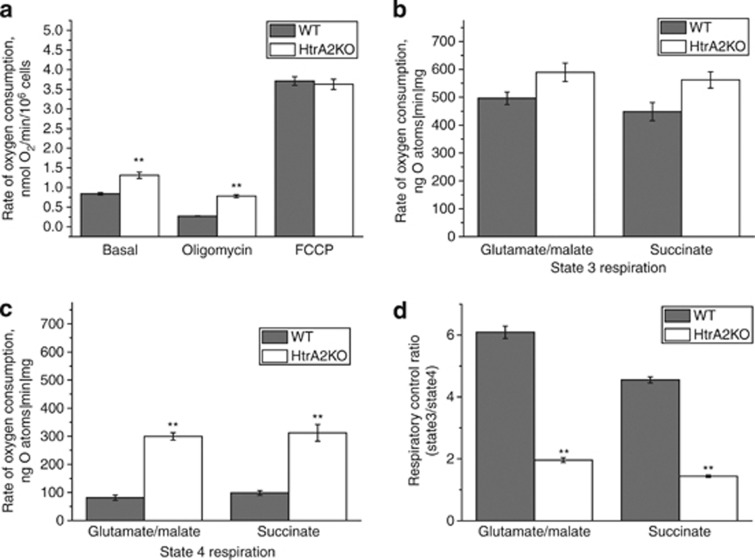
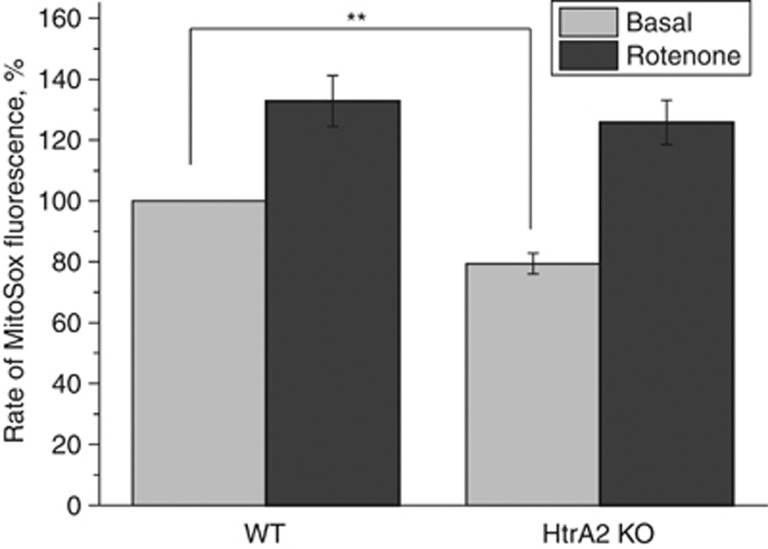
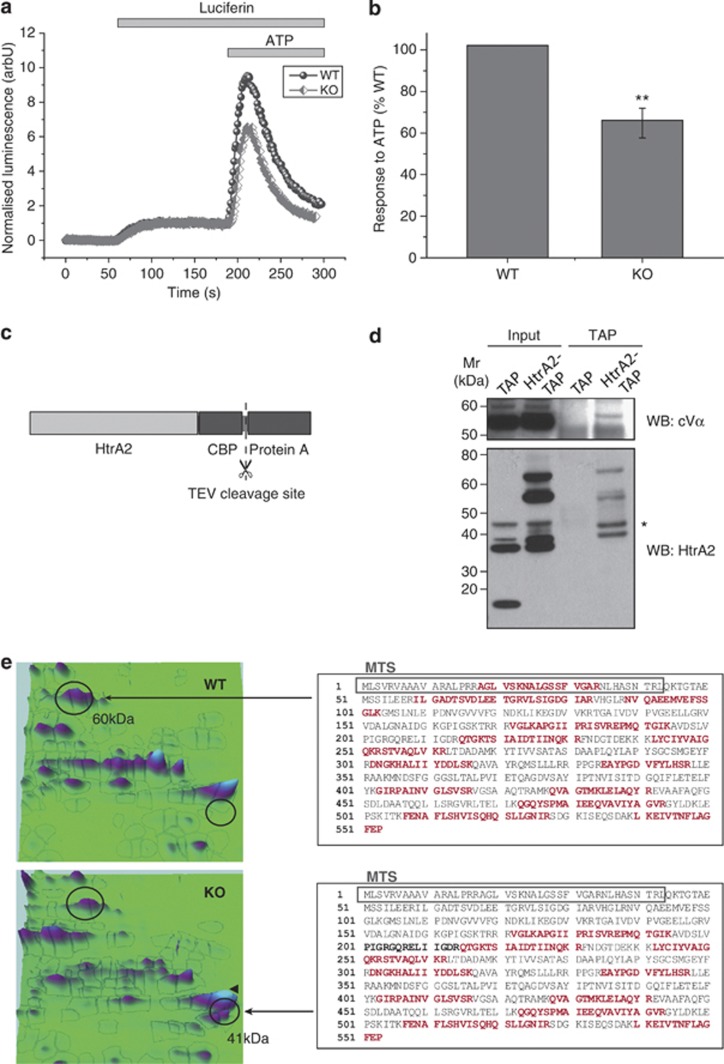
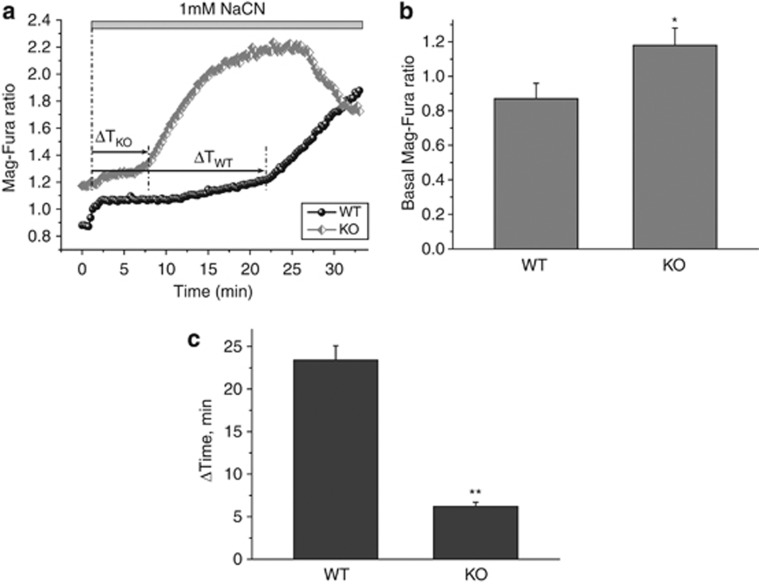
Loss of the mitochondrial protease HtrA2 (Omi) in mice leads to mitochondrial dysfunction, neurodegeneration and premature death, but the mechanism underlying this pathology remains unclear. Using primary cultures from wild-type and HtrA2-knockout mice, we find that HtrA2 deficiency significantly reduces mitochondrial membrane potential in a range of cell types. This depolarisation was found to result from mitochondrial uncoupling, as mitochondrial respiration was increased in HtrA2-deficient cells and respiratory control ratio was dramatically reduced. HtrA2-knockout cells exhibit increased proton translocation through the ATP synthase, in combination with decreased ATP production and truncation of the F1 α-subunit, suggesting the ATP synthase as the source of the proton leak. Uncoupling in the HtrA2-deficient mice is accompanied by altered breathing pattern and, on a cellular level, ATP depletion and vulnerability to chemical ischaemia. We propose that this vulnerability may ultimately cause the neurodegeneration observed in these mice.
线粒体蛋白酶 HtrA2(Omi)缺失的小鼠会导致线粒体功能障碍、神经退行性变和早逝,但这种病变的机制仍不清楚。使用来自野生型和 HtrA2 敲除小鼠的原代培养物,我们发现 HtrA2 缺乏会显著降低多种细胞类型中线粒体膜电位。这种去极化被发现是由于线粒体解偶联引起的,因为 HtrA2 缺失细胞中的线粒体呼吸增加,呼吸控制比显著降低。HtrA2 敲除细胞表现出通过 ATP 合酶的质子转运增加,同时伴随着 ATP 生成减少和 F1α亚基截断,表明 ATP 合酶是质子漏的来源。在 HtrA2 缺失的小鼠中,解偶联伴随着呼吸模式的改变,在细胞水平上,ATP 耗竭和对化学缺氧的脆弱性。我们提出,这种脆弱性可能最终导致这些小鼠中观察到的神经退行性变。