Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX, USA.
BMC Microbiol. 2012 Jul 7;12:135. doi: 10.1186/1471-2180-12-135.
Enterococci are among the leading causes of hospital-acquired infections in the United States and Europe, with Enterococcus faecalis and Enterococcus faecium being the two most common species isolated from enterococcal infections. In the last decade, the proportion of enterococcal infections caused by E. faecium has steadily increased compared to other Enterococcus species. Although the underlying mechanism for the gradual replacement of E. faecalis by E. faecium in the hospital environment is not yet understood, many studies using genotyping and phylogenetic analysis have shown the emergence of a globally dispersed polyclonal subcluster of E. faecium strains in clinical environments. Systematic study of the molecular epidemiology and pathogenesis of E. faecium has been hindered by the lack of closed, complete E. faecium genomes that can be used as references.
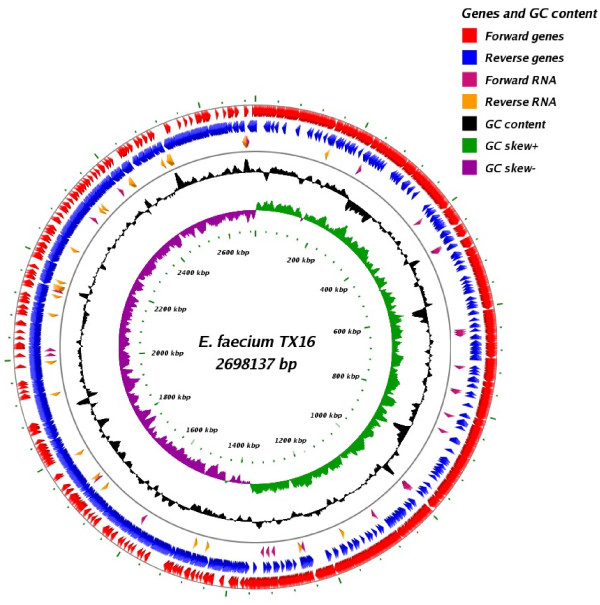
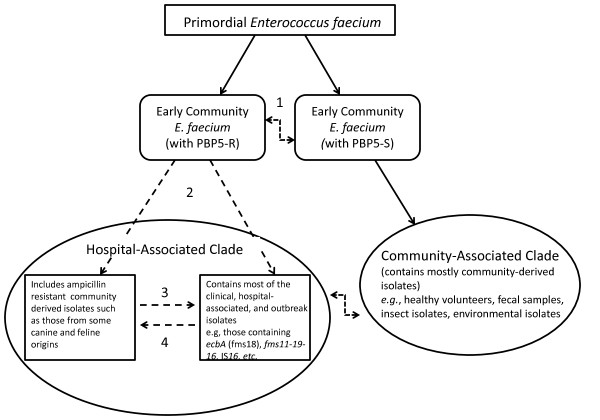
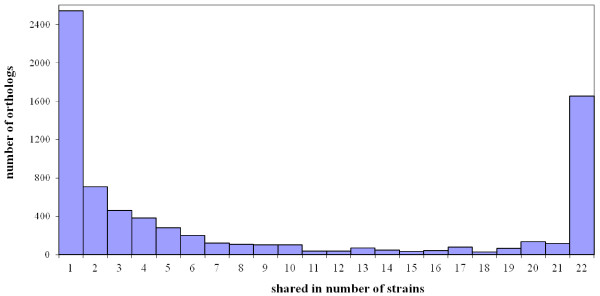
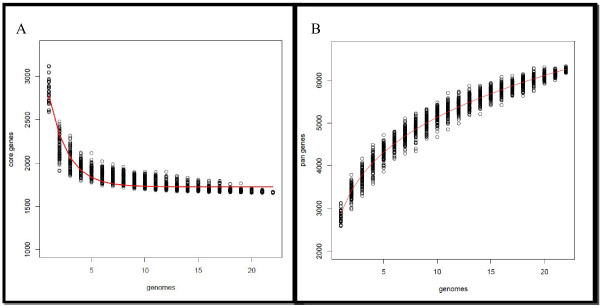
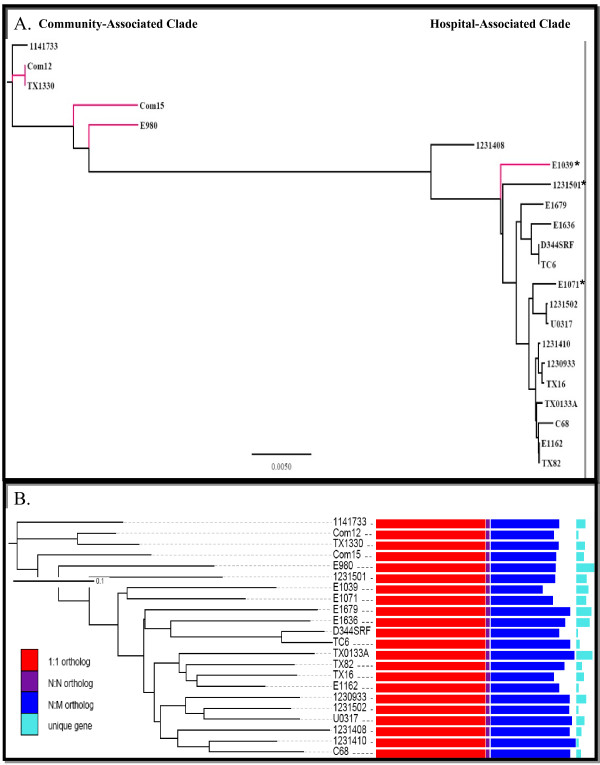
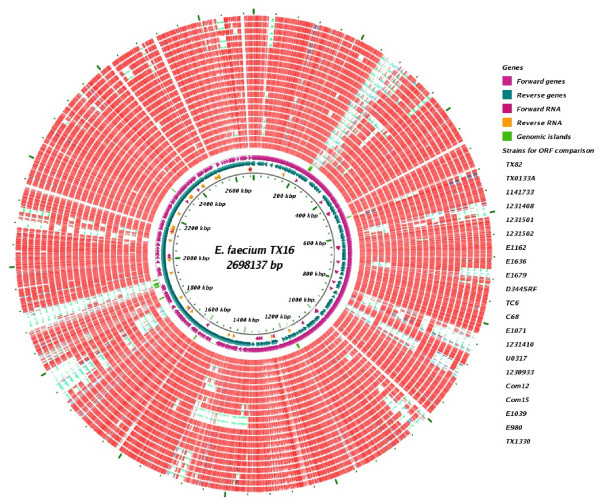
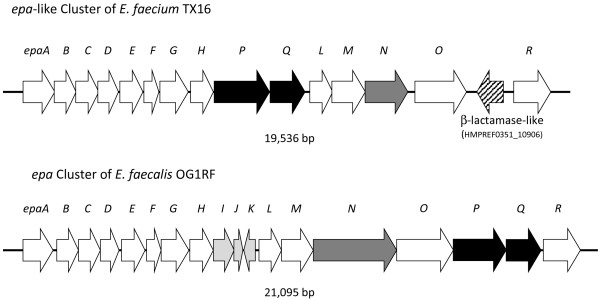
In this study, we report the complete genome sequence of the E. faecium strain TX16, also known as DO, which belongs to multilocus sequence type (ST) 18, and was the first E. faecium strain ever sequenced. Whole genome comparison of the TX16 genome with 21 E. faecium draft genomes confirmed that most clinical, outbreak, and hospital-associated (HA) strains (including STs 16, 17, 18, and 78), in addition to strains of non-hospital origin, group in the same clade (referred to as the HA clade) and are evolutionally considerably more closely related to each other by phylogenetic and gene content similarity analyses than to isolates in the community-associated (CA) clade with approximately a 3-4% average nucleotide sequence difference between the two clades at the core genome level. Our study also revealed that many genomic loci in the TX16 genome are unique to the HA clade. 380 ORFs in TX16 are HA-clade specific and antibiotic resistance genes are enriched in HA-clade strains. Mobile elements such as IS16 and transposons were also found almost exclusively in HA strains, as previously reported.
Our findings along with other studies show that HA clonal lineages harbor specific genetic elements as well as sequence differences in the core genome which may confer selection advantages over the more heterogeneous CA E. faecium isolates. Which of these differences are important for the success of specific E. faecium lineages in the hospital environment remain(s) to be determined.
肠球菌是美国和欧洲医院获得性感染的主要原因之一,其中粪肠球菌和屎肠球菌是从肠球菌感染中分离出来的两种最常见的物种。在过去的十年中,与其他肠球菌物种相比,粪肠球菌引起的肠球菌感染比例稳步增加。尽管医院环境中粪肠球菌逐渐被屎肠球菌取代的潜在机制尚不清楚,但许多使用基因分型和系统发育分析的研究表明,临床环境中出现了一种具有全球分布的屎肠球菌多克隆亚群。由于缺乏可作为参考的封闭、完整的屎肠球菌基因组,对屎肠球菌的分子流行病学和发病机制进行系统研究受到了阻碍。
在这项研究中,我们报告了屎肠球菌菌株 TX16 的完整基因组序列,该菌株也称为 DO,属于多位序列型(ST)18,是第一个被测序的屎肠球菌菌株。TX16 基因组与 21 个屎肠球菌草图基因组的全基因组比较证实,大多数临床、暴发和医院相关(HA)菌株(包括 ST16、17、18 和 78),以及非医院来源的菌株,都聚集在同一个进化枝(称为 HA 进化枝),并且通过系统发育和基因内容相似性分析,彼此之间的进化关系比社区相关(CA)进化枝更为密切,两个进化枝在核心基因组水平上的平均核苷酸序列差异约为 3-4%。我们的研究还表明,TX16 基因组中的许多基因组基因座是 HA 进化枝特有的。TX16 中有 380 个 ORF 是 HA 进化枝特有的,抗生素耐药基因在 HA 进化枝菌株中富集。像以前报道的那样,IS16 和转座子等移动元件也几乎只存在于 HA 菌株中。
我们的研究结果以及其他研究表明,HA 克隆谱系携带有特定的遗传元件以及核心基因组中的序列差异,这可能赋予其相对于更具异质性的 CA 屎肠球菌分离株的选择优势。这些差异中哪些对特定屎肠球菌谱系在医院环境中的成功至关重要,还有待确定。