Bakan Ahmet, Nevins Neysa, Lakdawala Ami S, Bahar Ivet
J Chem Theory Comput. 2012 Jul 10;8(7):2435-2447. doi: 10.1021/ct300117j. Epub 2012 Jun 5.
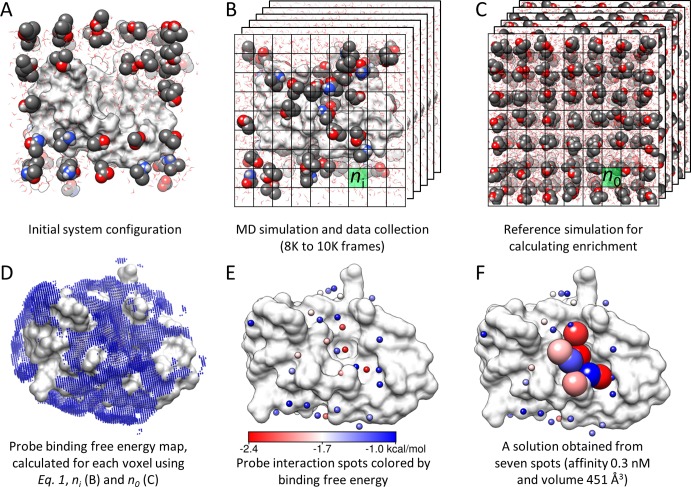
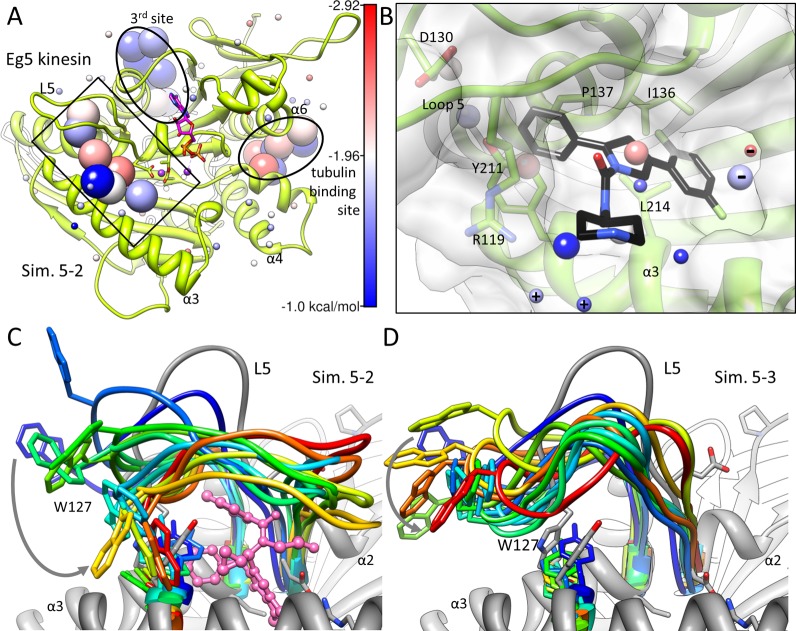
Druggability assessment of a target protein has emerged in recent years as an important concept in hit-to-lead optimization. A reliable and physically relevant measure of druggability would allow informed decisions on the risk of investing in a particular target. Here, we define "druggability" as a quantitative estimate of binding sites and affinities for a potential drug acting on a specific protein target. In the present study, we describe a new methodology that successfully predicts the druggability and maximal binding affinity for a series of challenging targets, including those that function through allosteric mechanisms. Two distinguishing features of the methodology are (i) simulation of the binding dynamics of a diversity of probe molecules selected on the basis of an analysis of approved drugs and (ii) identification of druggable sites and estimation of corresponding binding affinities on the basis of an evaluation of the geometry and energetics of bound probe clusters. The use of the methodology for a variety of targets such as murine double mutant-2, protein tyrosine phosphatase 1B (PTP1B), lymphocyte function-associated antigen 1, vertebrate kinesin-5 (Eg5), and p38 mitogen-activated protein kinase provides examples for which the method correctly captures the location and binding affinities of known drugs. It also provides insights into novel druggable sites and the target's structural changes that would accommodate, if not promote and stabilize, drug binding. Notably, the ability to identify high affinity spots even in challenging cases such as PTP1B or Eg5 shows promise as a rational tool for assessing the druggability of protein targets and identifying allosteric or novel sites for drug binding.
近年来,靶蛋白的成药潜力评估已成为从苗头化合物到先导化合物优化过程中的一个重要概念。一种可靠且与物理相关的成药潜力衡量标准将有助于就投资特定靶点的风险做出明智决策。在此,我们将“成药潜力”定义为作用于特定蛋白质靶点的潜在药物的结合位点和亲和力的定量估计。在本研究中,我们描述了一种新方法,该方法成功预测了一系列具有挑战性的靶点的成药潜力和最大结合亲和力,包括那些通过变构机制发挥作用的靶点。该方法的两个显著特点是:(i)基于对已批准药物的分析选择多种探针分子,并模拟其结合动力学;(ii)基于对结合探针簇的几何结构和能量学的评估,识别可成药位点并估计相应的结合亲和力。将该方法应用于多种靶点,如小鼠双突变体-2、蛋白酪氨酸磷酸酶1B(PTP1B)、淋巴细胞功能相关抗原1、脊椎动物驱动蛋白-5(Eg5)和p38丝裂原活化蛋白激酶,这些例子表明该方法能够正确捕捉已知药物的位置和结合亲和力。它还为新型可成药位点以及靶点的结构变化提供了见解,这些结构变化即使不能促进和稳定药物结合,也能容纳药物结合。值得注意的是,即使在PTP1B或Eg5等具有挑战性的情况下,该方法识别高亲和力位点的能力也显示出有望成为评估蛋白质靶点成药潜力和识别变构或新型药物结合位点的合理工具。