Department of Computational and Systems Biology, and Clinical & Translational Science Institute, School of Medicine, University of Pittsburgh, Pittsburgh, PA 15213, USA.
Bioinformatics. 2011 Jun 1;27(11):1575-7. doi: 10.1093/bioinformatics/btr168. Epub 2011 Apr 5.
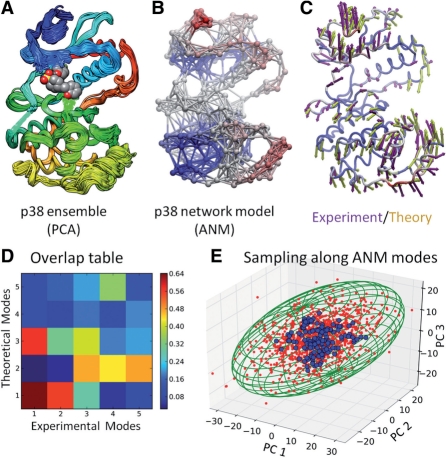
We developed a Python package, ProDy, for structure-based analysis of protein dynamics. ProDy allows for quantitative characterization of structural variations in heterogeneous datasets of structures experimentally resolved for a given biomolecular system, and for comparison of these variations with the theoretically predicted equilibrium dynamics. Datasets include structural ensembles for a given family or subfamily of proteins, their mutants and sequence homologues, in the presence/absence of their substrates, ligands or inhibitors. Numerous helper functions enable comparative analysis of experimental and theoretical data, and visualization of the principal changes in conformations that are accessible in different functional states. ProDy application programming interface (API) has been designed so that users can easily extend the software and implement new methods.
ProDy is open source and freely available under GNU General Public License from http://www.csb.pitt.edu/ProDy/.
我们开发了一个名为 ProDy 的 Python 包,用于基于结构的蛋白质动力学分析。ProDy 允许对给定生物分子系统的实验解析结构的异质数据集的结构变化进行定量描述,并将这些变化与理论预测的平衡动力学进行比较。数据集包括给定蛋白质家族或亚家族的结构集合、它们的突变体和序列同源物,以及在存在/不存在其底物、配体或抑制剂的情况下。许多辅助功能可实现实验和理论数据的比较分析,并可视化在不同功能状态下可及的构象的主要变化。ProDy 的应用程序编程接口 (API) 设计为用户可以轻松扩展软件并实现新方法。
ProDy 是开源的,根据 GNU 通用公共许可证可免费从 http://www.csb.pitt.edu/ProDy/ 获取。