Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota, Minneapolis, Minnesota, United States of America.
PLoS Pathog. 2012;8(7):e1002800. doi: 10.1371/journal.ppat.1002800. Epub 2012 Jul 12.
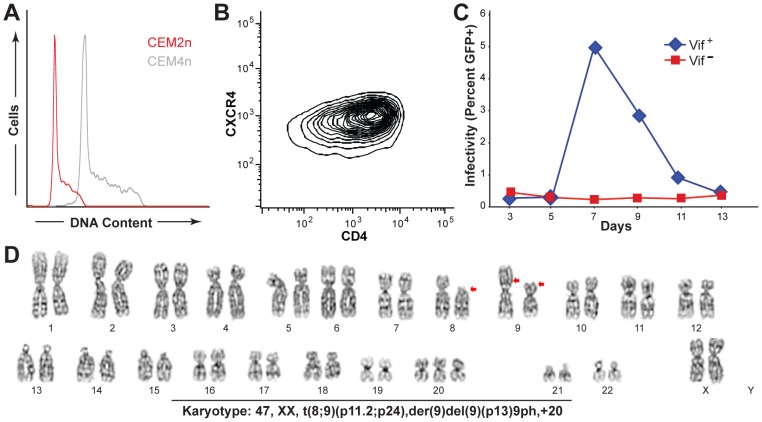
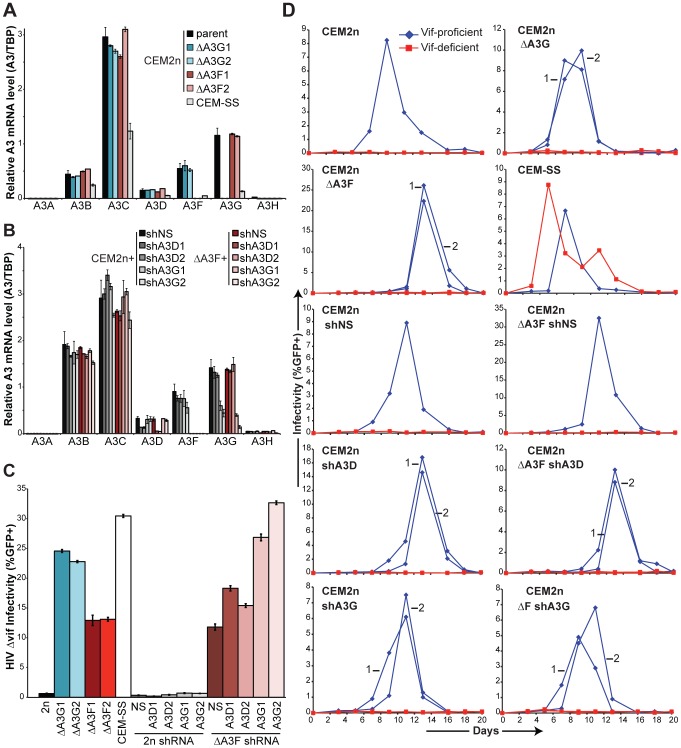
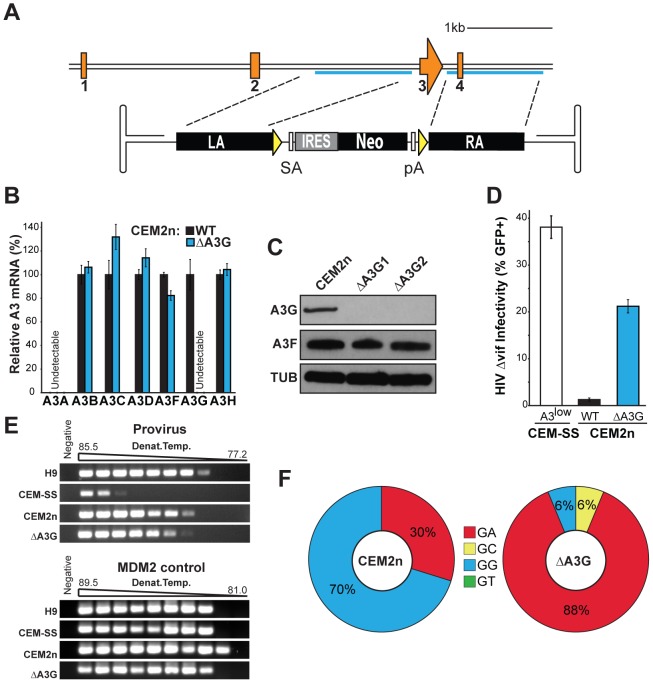
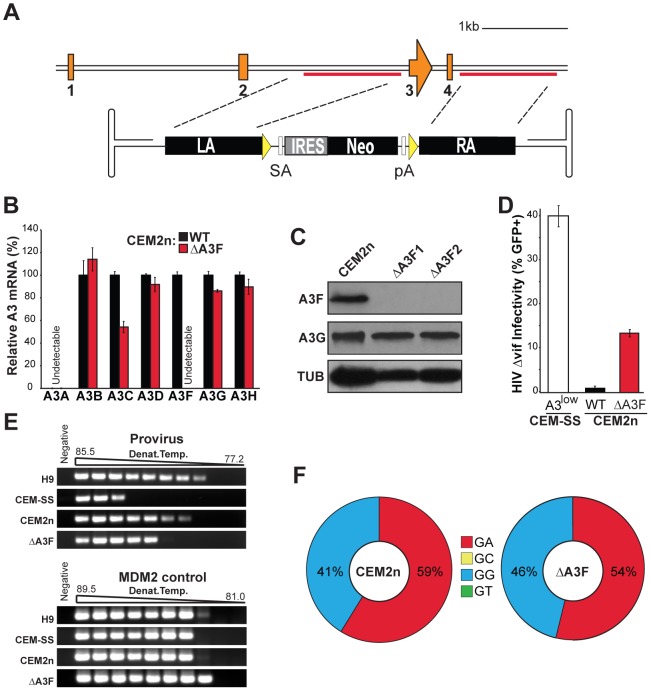
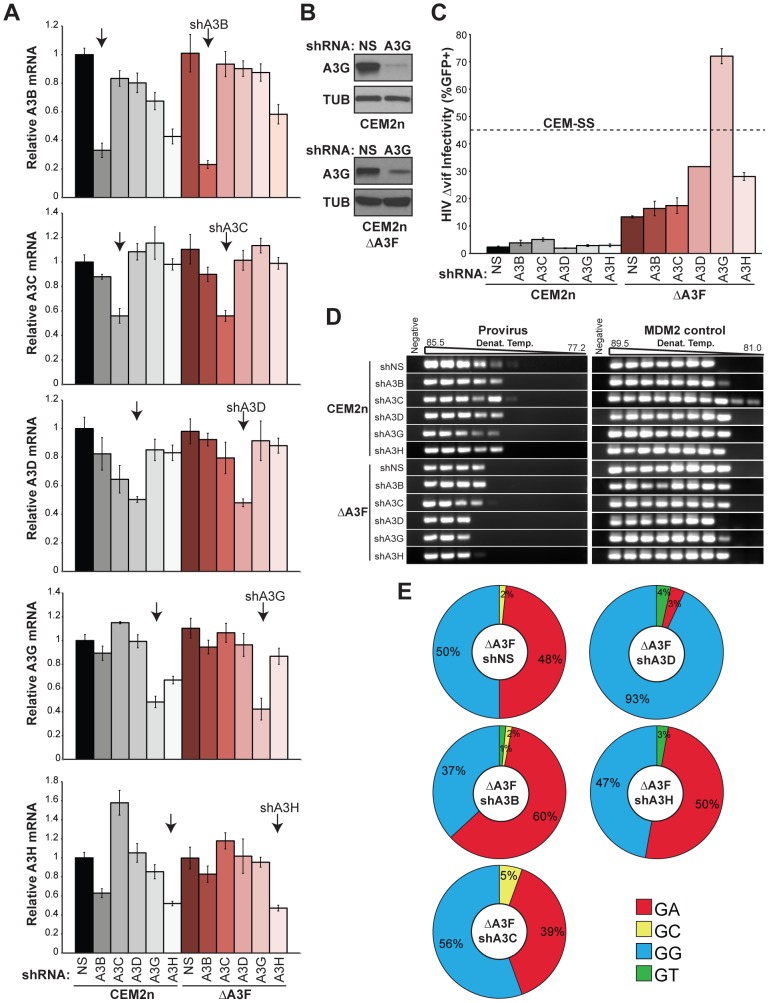
The DNA deaminase APOBEC3G converts cytosines to uracils in retroviral cDNA, which are immortalized as genomic strand G-to-A hypermutations by reverse transcription. A single round of APOBEC3G-dependent mutagenesis can be catastrophic, but evidence suggests that sublethal levels contribute to viral genetic diversity and the associated problems of drug resistance and immune escape. APOBEC3G exhibits an intrinsic preference for the second cytosine in a 5'CC dinucleotide motif leading to 5'GG-to-AG mutations. However, an additional hypermutation signature is commonly observed in proviral sequences from HIV-1 infected patients, 5'GA-to-AA, and it has been attributed controversially to one or more of the six other APOBEC3 deaminases. An unambiguous resolution of this problem has been difficult to achieve, in part due to dominant effects of protein over-expression. Here, we employ gene targeting to dissect the endogenous APOBEC3 contribution to Vif-deficient HIV-1 restriction and hypermutation in a nonpermissive T cell line CEM2n. We report that APOBEC3G-null cells, as predicted from previous studies, lose the capacity to inflict 5'GG-to-AG mutations. In contrast, APOBEC3F-null cells produced viruses with near-normal mutational patterns. Systematic knockdown of other APOBEC3 genes in an APOBEC3F-null background revealed a significant contribution from APOBEC3D in promoting 5'GA-to-AA hypermutations. Furthermore, Vif-deficient HIV-1 restriction was strong in parental CEM2n and APOBEC3D-knockdown cells, partially alleviated in APOBEC3G- or APOBEC3F-null cells, further alleviated in APOBEC3F-null/APOBEC3D-knockdown cells, and alleviated to the greatest extent in APOBEC3F-null/APOBEC3G-knockdown cells revealing clear redundancy in the HIV-1 restriction mechanism. We conclude that endogenous levels of APOBEC3D, APOBEC3F, and APOBEC3G combine to restrict Vif-deficient HIV-1 and cause the hallmark dinucleotide hypermutation patterns in CEM2n. Primary T lymphocytes express a similar set of APOBEC3 genes suggesting that the same repertoire may be important in vivo.
APOBEC3G 是一种 DNA 脱氨酶,可将逆转录病毒 cDNA 中的胞嘧啶转化为尿嘧啶,从而将其固定为基因组链的 G 到 A 超突变。APOBEC3G 依赖性诱变的单个循环可能是灾难性的,但有证据表明,亚致死水平有助于病毒遗传多样性,并导致耐药性和免疫逃逸等相关问题。APOBEC3G 对 5'CC 二核苷酸基序中的第二个胞嘧啶表现出内在的偏好,导致 5'GG 到 AG 突变。然而,在感染 HIV-1 的患者的前病毒序列中通常观察到另一种超突变特征,即 5'GA 到 AA,并且有争议地归因于六种其他 APOBEC3 脱氨酶之一或多种。由于蛋白过表达的主导作用,这个问题的明确解决一直很困难。在这里,我们采用基因靶向技术来剖析内源性 APOBEC3 对 Vif 缺陷型 HIV-1 限制和 CEM2n 非许可性 T 细胞系中的超突变的贡献。我们报告说,正如先前的研究预测的那样,APOBEC3G 缺失细胞丧失了施加 5'GG 到 AG 突变的能力。相比之下,APOBEC3F 缺失细胞产生的病毒具有近乎正常的突变模式。在 APOBEC3F 缺失背景下系统敲低其他 APOBEC3 基因表明,APOBEC3D 显著促进 5'GA 到 AA 超突变。此外,Vif 缺陷型 HIV-1 在亲本 CEM2n 和 APOBEC3D 敲低细胞中的限制很强,在 APOBEC3G 或 APOBEC3F 缺失细胞中部分缓解,在 APOBEC3F 缺失/APOBEC3D 敲低细胞中进一步缓解,在 APOBEC3F 缺失/APOBEC3G 敲低细胞中缓解到最大程度,表明 HIV-1 限制机制存在明显的冗余。我们得出结论,内源性水平的 APOBEC3D、APOBEC3F 和 APOBEC3G 共同限制 Vif 缺陷型 HIV-1 并导致 CEM2n 中标志性的二核苷酸超突变模式。原代 T 淋巴细胞表达相似的 APOBEC3 基因集,表明相同的基因库在体内可能很重要。