Hao Ming, Ren Hong, Luo Fang, Zhang Shuwei, Qiu Jieshan, Ji Mingjuan, Si Hongzong, Li Guohui
Department of Materials Science and Chemical Engineering, Dalian University of Technology, Dalian 116023, China.
Department of Ophthalmology, Qi Lu Hospital, Medical School of Shandong University, Jinan 250012, China.
Int J Mol Sci. 2012;13(6):7057-7079. doi: 10.3390/ijms13067057. Epub 2012 Jun 8.
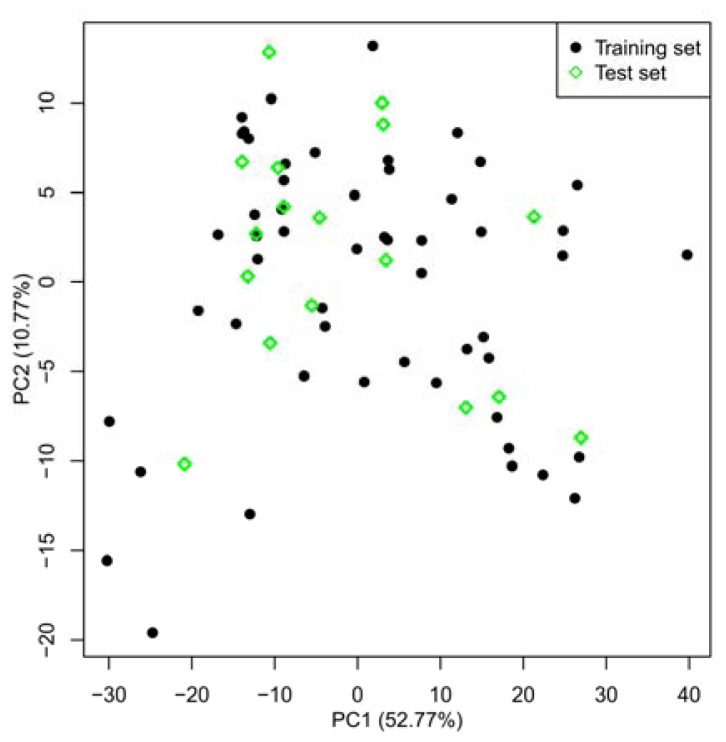
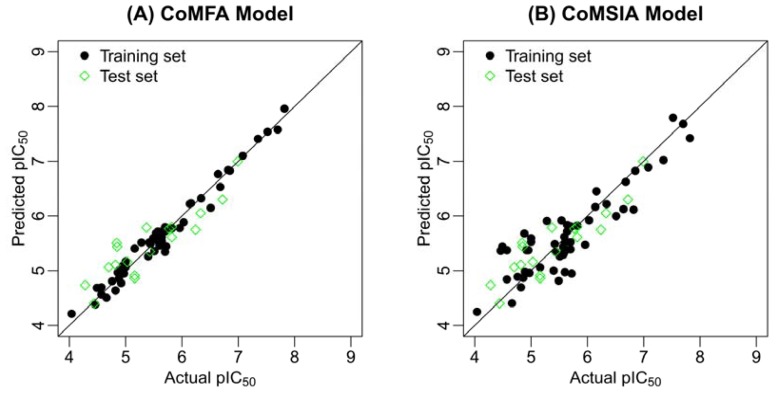
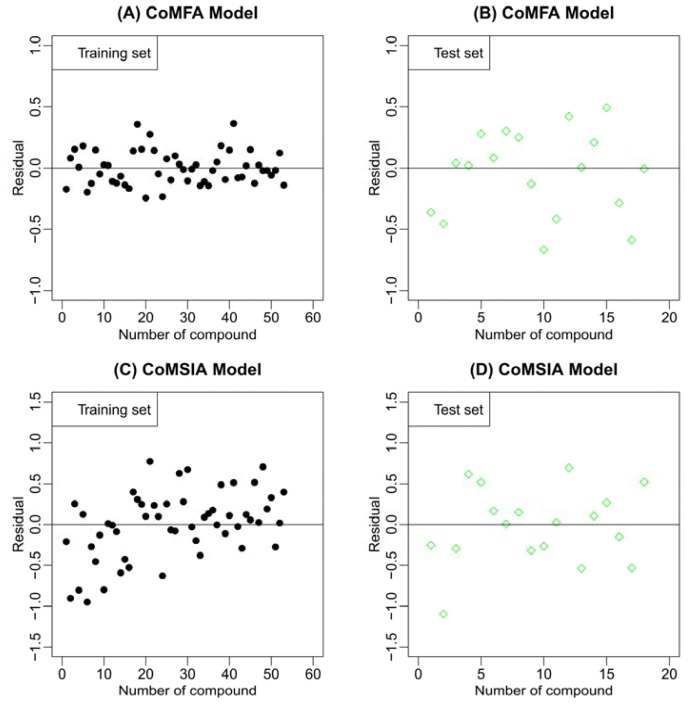
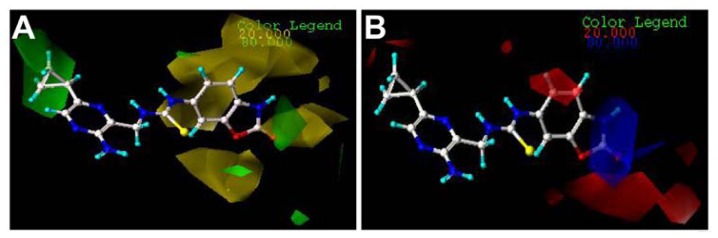
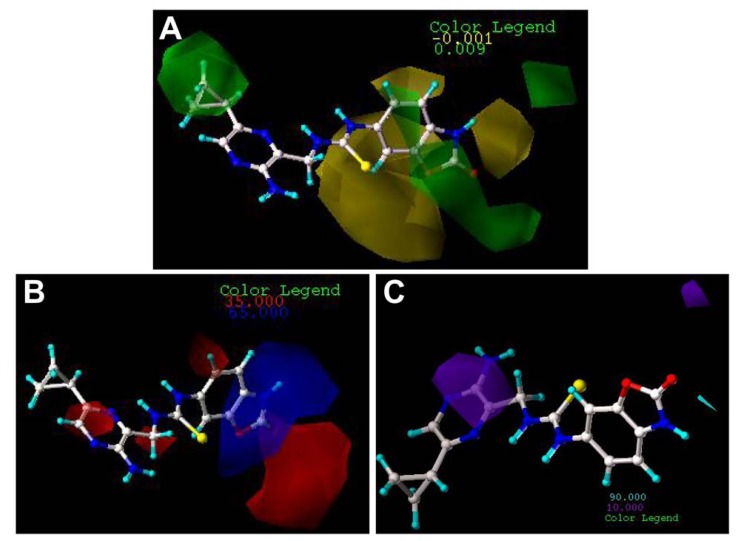
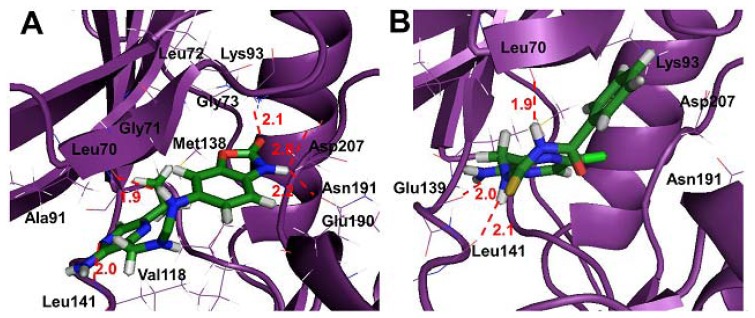
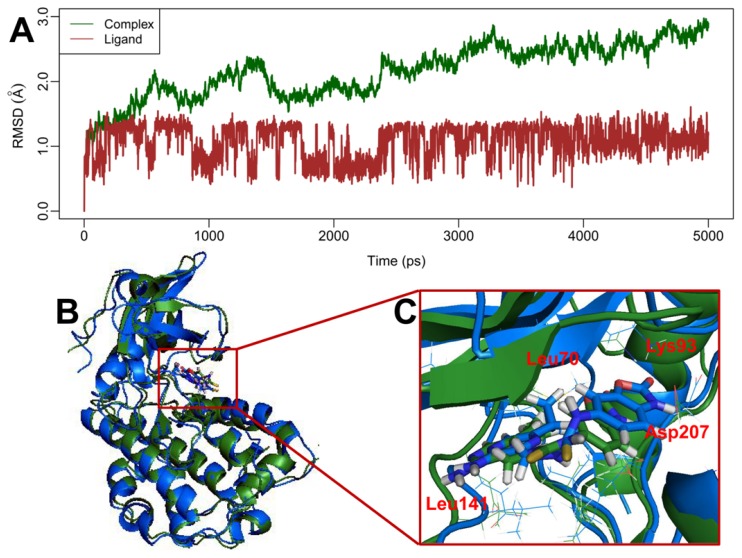
Mitogen-activated protein kinase-activated protein kinase 2 (MK-2) has been identified as a drug target for the treatment of inflammatory diseases. Currently, a series of thiourea analogs as potent MK-2 inhibitors were studied using comprehensive computational methods by 3D-QSAR, molecular docking and molecular dynamics simulations for a further improvement in activities. The optimal 3D models exhibit high statistical significance of the results, especially for the CoMFA results with r(2) (ncv), q(2) values of 0.974, 0.536 for the internal validation, and r(2) (pred), r(2) (m) values of 0.910, 0.723 for the external validation and Roy's index, respectively. In addition, more rigorous validation criteria suggested by Tropsha were also employed to check the built models. Graphic representation of the results, as contoured 3D coefficient plots, also provides a clue to the reasonable modification of molecules: (i) The substituent with a bulky size and electron-rich group at the C5 position of the pyrazine ring is required to enhance the potency; (ii) The H-bond acceptor group in the C3 position of the pyrazine ring is likely to be helpful to increase MK-2 inhibition; (iii) The small and electropositive substituent as a hydrogen bond donor of the C2 position in the oxazolone ring is favored; In addition, several important amino acid residues were also identified as playing an important role in MK-2 inhibition. The agreement between 3D-QSAR, molecular docking and molecular dynamics simulations also proves the rationality of the developed models. These results, we hope, may be helpful in designing novel and potential MK-2 inhibitors.
丝裂原活化蛋白激酶激活的蛋白激酶2(MK-2)已被确定为治疗炎症性疾病的药物靶点。目前,通过3D-QSAR、分子对接和分子动力学模拟等综合计算方法,对一系列作为强效MK-2抑制剂的硫脲类似物进行了研究,以进一步提高其活性。优化后的3D模型结果具有较高的统计学意义,特别是对于CoMFA结果,内部验证的r(2)(ncv)、q(2)值分别为0.974、0.536,外部验证的r(2)(pred)、r(2)(m)值分别为0.910、0.723以及Roy指数。此外,还采用了Tropsha提出的更严格的验证标准来检验所构建的模型。结果的图形表示,如等高线3D系数图,也为分子的合理修饰提供了线索:(i)吡嗪环C5位需要具有大体积和富电子基团的取代基来增强效力;(ii)吡嗪环C3位的氢键受体基团可能有助于增加对MK-2的抑制作用;(iii)恶唑酮环C2位作为氢键供体的小的、带正电的取代基是有利的;此外,还确定了几个重要的氨基酸残基在MK-2抑制中起重要作用。3D-QSAR、分子对接和分子动力学模拟之间的一致性也证明了所开发模型的合理性。我们希望这些结果可能有助于设计新型且有潜力的MK-2抑制剂。