Pettengill James B, McAvoy Eugene, White James R, Allard Marc, Brown Eric, Ottesen Andrea
FDA Center for Food Safety and Applied Nutrition, Division of Microbiology, Molecular Methods and Subtyping, College Park, MD 20740, USA.
BMC Res Notes. 2012 Jul 27;5:378. doi: 10.1186/1756-0500-5-378.
Enriching environmental samples to increase the probability of detection has been standard practice throughout the history of microbiology. However, by its very nature, the process of enrichment creates a biased sample that may have unintended consequences for surveillance or resolving a pathogenic outbreak. With the advent of next-generation sequencing and metagenomic approaches, the possibility now exists to quantify enrichment bias at an unprecedented taxonomic breadth.
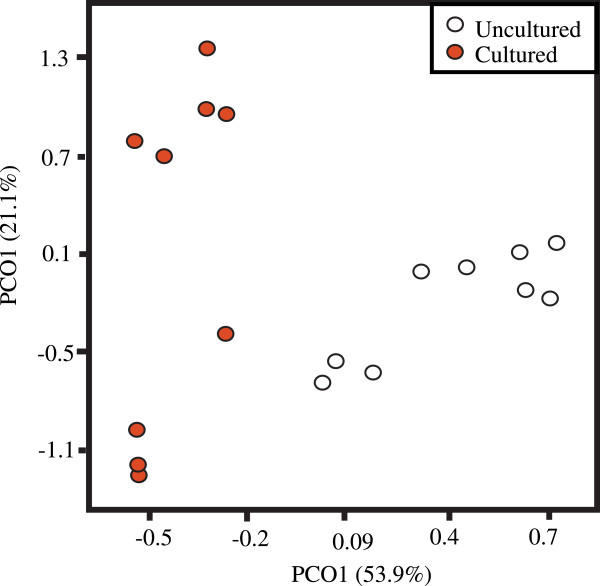
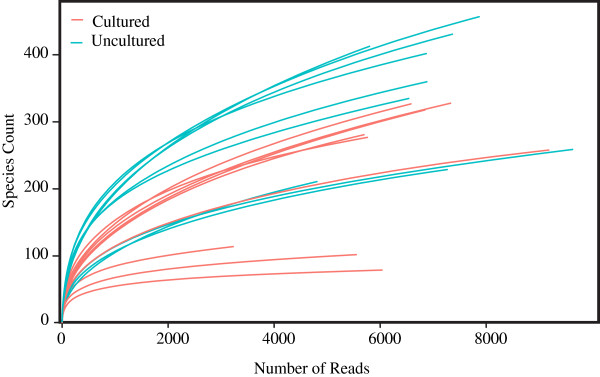
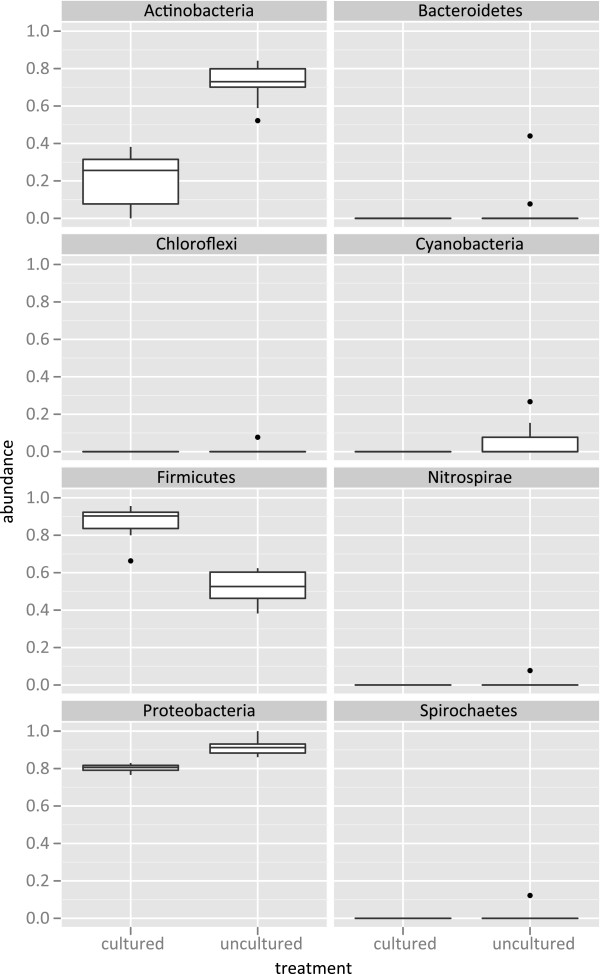
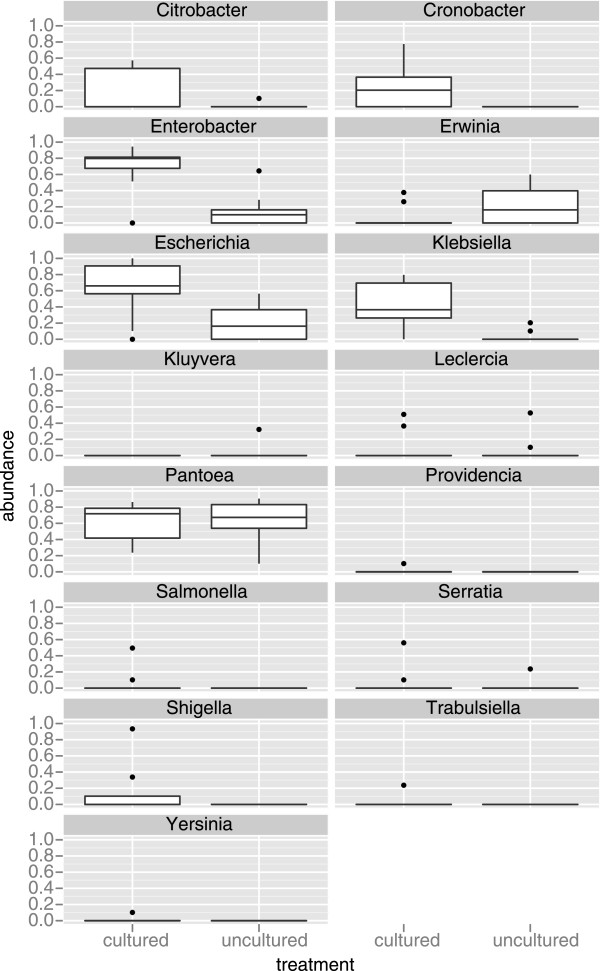
We investigated differences in taxonomic profiles of three enriched and unenriched tomato phyllosphere samples taken from three different tomato fields (n = 18). 16S rRNA gene meteganomes were created for each of the 18 samples using 454/Roche's pyrosequencing platform, resulting in a total of 165,259 sequences. Significantly different taxonomic profiles and abundances at a number of taxonomic levels were observed between the two treatments. Although as many as 28 putative Salmonella sequences were detected in enriched samples, there was no significant difference in the abundance of Salmonella between enriched and unenriched treatments.
Our results illustrate that the process of enriching greatly alters the taxonomic profile of an environmental sample beyond that of the target organism. We also found evidence suggesting that enrichment may not increase the probability of detecting a target. In conclusion, our results further emphasize the need to develop metagenomics as a validated culture independent method for pathogen detection.
在微生物学发展历程中,富集环境样本以提高检测概率一直是标准做法。然而,富集过程本身会产生有偏差的样本,这可能给监测或解决致病性疫情带来意想不到的后果。随着新一代测序和宏基因组学方法的出现,现在有机会以前所未有的分类广度对富集偏差进行量化。
我们调查了从三个不同番茄田采集的三个富集和未富集番茄叶际样本(n = 18)的分类学特征差异。使用454/罗氏焦磷酸测序平台为18个样本中的每个样本创建16S rRNA基因宏基因组,共产生165,259条序列。在两种处理之间,在多个分类水平上观察到显著不同的分类学特征和丰度。虽然在富集样本中检测到多达28条推测的沙门氏菌序列,但富集处理和未富集处理之间沙门氏菌的丰度没有显著差异。
我们的结果表明,富集过程极大地改变了环境样本的分类学特征,超出了目标生物体的分类学特征。我们还发现有证据表明富集可能不会增加检测到目标的概率。总之,我们的结果进一步强调了将宏基因组学发展成为一种经过验证的、独立于培养的病原体检测方法的必要性。