Department of Genetics, Development, and Cell Biology, Iowa State University, Ames, Iowa 50011, USA.
BMC Bioinformatics. 2012 Aug 1;13:187. doi: 10.1186/1471-2105-13-187.
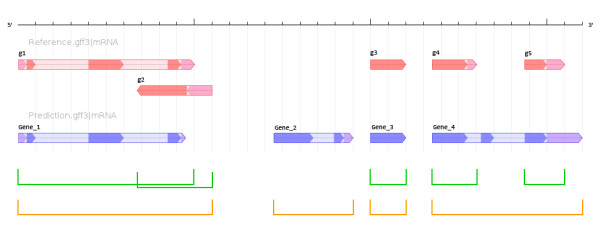
Accurate gene structure annotation is a fundamental but somewhat elusive goal of genome projects, as witnessed by the fact that (model) genomes typically undergo several cycles of re-annotation. In many cases, it is not only different versions of annotations that need to be compared but also different sources of annotation of the same genome, derived from distinct gene prediction workflows. Such comparisons are of interest to annotation providers, prediction software developers, and end-users, who all need to assess what is common and what is different among distinct annotation sources. We developed ParsEval, a software application for pairwise comparison of sets of gene structure annotations. ParsEval calculates several statistics that highlight the similarities and differences between the two sets of annotations provided. These statistics are presented in an aggregate summary report, with additional details provided as individual reports specific to non-overlapping, gene-model-centric genomic loci. Genome browser styled graphics embedded in these reports help visualize the genomic context of the annotations. Output from ParsEval is both easily read and parsed, enabling systematic identification of problematic gene models for subsequent focused analysis.
ParsEval is capable of analyzing annotations for large eukaryotic genomes on typical desktop or laptop hardware. In comparison to existing methods, ParsEval exhibits a considerable performance improvement, both in terms of runtime and memory consumption. Reports from ParsEval can provide relevant biological insights into the gene structure annotations being compared.
Implemented in C, ParsEval provides the quickest and most feature-rich solution for genome annotation comparison to date. The source code is freely available (under an ISC license) at http://parseval.sourceforge.net/.
准确的基因结构注释是基因组计划的基本但有些难以捉摸的目标,这从(模型)基因组通常需要经历几个周期的重新注释就可以看出。在许多情况下,不仅需要比较不同版本的注释,还需要比较同一基因组的不同注释来源,这些来源来自不同的基因预测工作流程。这些比较对于注释提供者、预测软件开发商和最终用户都很感兴趣,他们都需要评估不同注释来源之间的共同点和不同点。我们开发了 ParsEval,这是一种用于基因结构注释集的两两比较的软件应用程序。ParsEval 计算了几个突出两个注释集之间相似性和差异性的统计数据。这些统计数据以汇总摘要报告的形式呈现,并为非重叠的、以基因模型为中心的基因组区域提供了特定的详细报告。这些报告中嵌入的基因组浏览器样式的图形有助于可视化注释的基因组上下文。ParsEval 的输出既易于阅读又易于解析,能够系统地识别有问题的基因模型,以便进行后续的重点分析。
ParsEval 能够在典型的桌面或笔记本电脑硬件上分析大型真核生物基因组的注释。与现有方法相比,ParsEval 在运行时间和内存消耗方面都有显著的性能提升。ParsEval 的报告可以为正在比较的基因结构注释提供相关的生物学见解。
ParsEval 用 C 语言实现,是迄今为止用于基因组注释比较的最快和功能最丰富的解决方案。源代码可在 http://parseval.sourceforge.net/ (根据 ISC 许可证)免费获得。