Zhu Yafeng, Engström Pär G, Tellgren-Roth Christian, Baudo Charles D, Kennell John C, Sun Sheng, Billmyre R Blake, Schröder Markus S, Andersson Anna, Holm Tina, Sigurgeirsson Benjamin, Wu Guangxi, Sankaranarayanan Sundar Ram, Siddharthan Rahul, Sanyal Kaustuv, Lundeberg Joakim, Nystedt Björn, Boekhout Teun, Dawson Thomas L, Heitman Joseph, Scheynius Annika, Lehtiö Janne
Science for Life Laboratory, Department of Oncology-Pathology, Karolinska Institutet, 17121 Solna, Sweden.
Science for Life Laboratory, Department of Biochemistry and Biophysics, Stockholm University, 17121 Solna, Sweden.
Nucleic Acids Res. 2017 Mar 17;45(5):2629-2643. doi: 10.1093/nar/gkx006.
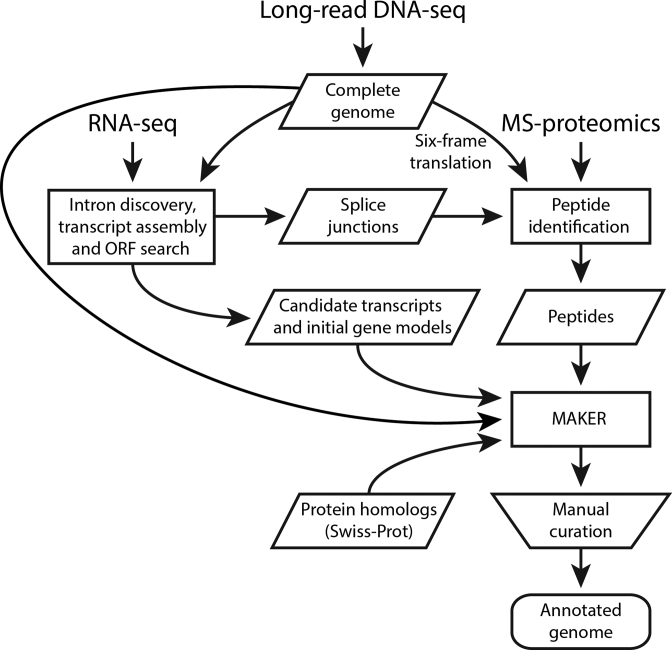
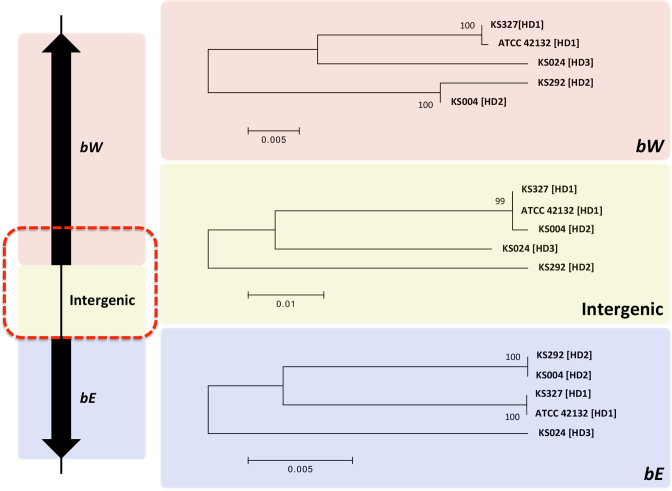
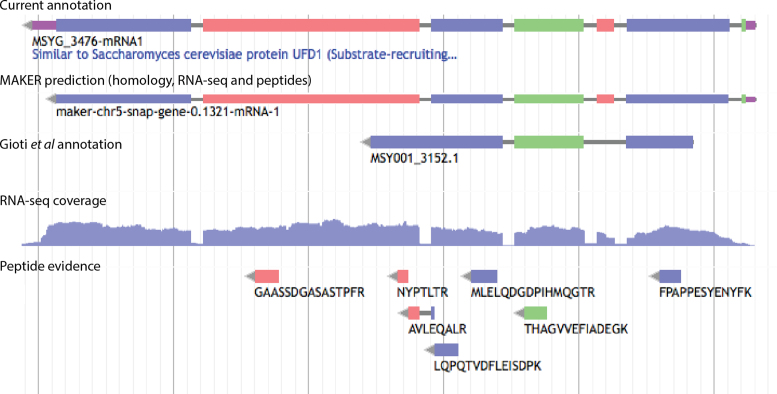
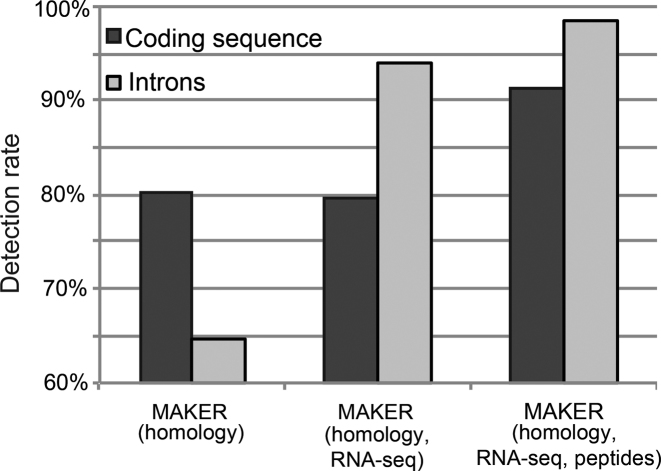
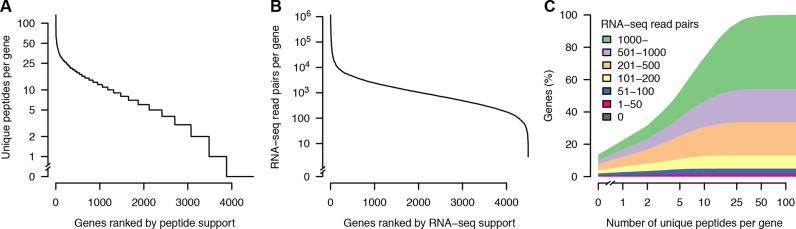
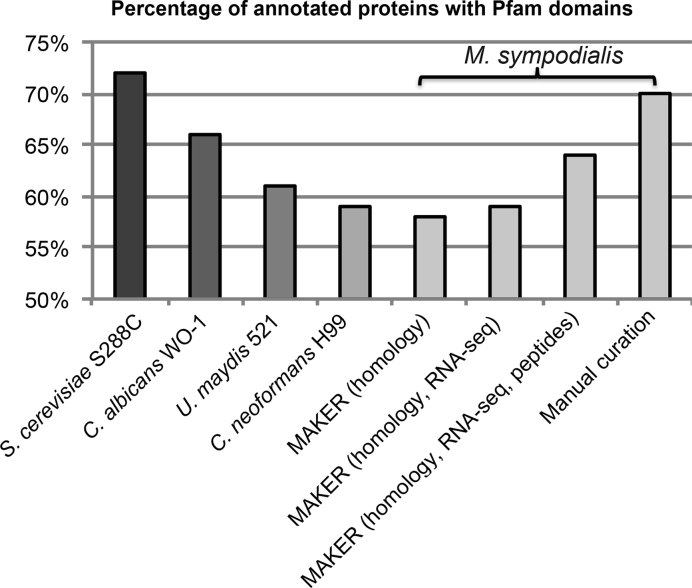
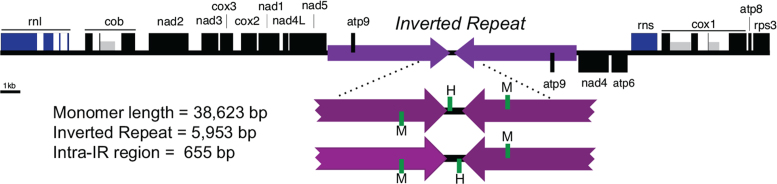
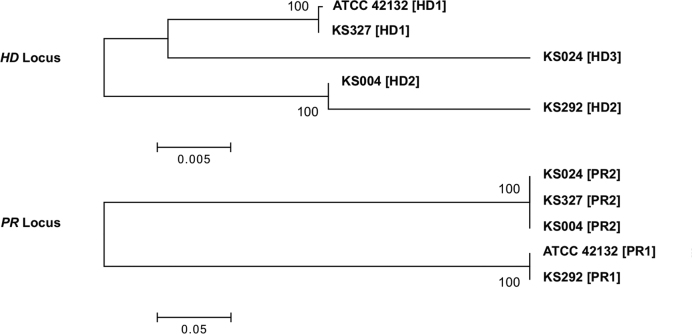
Complete and accurate genome assembly and annotation is a crucial foundation for comparative and functional genomics. Despite this, few complete eukaryotic genomes are available, and genome annotation remains a major challenge. Here, we present a complete genome assembly of the skin commensal yeast Malassezia sympodialis and demonstrate how proteogenomics can substantially improve gene annotation. Through long-read DNA sequencing, we obtained a gap-free genome assembly for M. sympodialis (ATCC 42132), comprising eight nuclear and one mitochondrial chromosome. We also sequenced and assembled four M. sympodialis clinical isolates, and showed their value for understanding Malassezia reproduction by confirming four alternative allele combinations at the two mating-type loci. Importantly, we demonstrated how proteomics data could be readily integrated with transcriptomics data in standard annotation tools. This increased the number of annotated protein-coding genes by 14% (from 3612 to 4113), compared to using transcriptomics evidence alone. Manual curation further increased the number of protein-coding genes by 9% (to 4493). All of these genes have RNA-seq evidence and 87% were confirmed by proteomics. The M. sympodialis genome assembly and annotation presented here is at a quality yet achieved only for a few eukaryotic organisms, and constitutes an important reference for future host-microbe interaction studies.
完整且准确的基因组组装和注释是比较基因组学和功能基因组学的关键基础。尽管如此,完整的真核生物基因组却很少见,基因组注释仍然是一项重大挑战。在此,我们展示了皮肤共生酵母合轴马拉色菌的完整基因组组装,并证明了蛋白质基因组学如何能显著改善基因注释。通过长读长DNA测序,我们获得了合轴马拉色菌(ATCC 42132)的无间隙基因组组装,其包含八条核染色体和一条线粒体染色体。我们还对四株合轴马拉色菌临床分离株进行了测序和组装,并通过确认两个交配型位点的四种替代等位基因组合,展示了它们在理解马拉色菌繁殖方面的价值。重要的是,我们展示了蛋白质组学数据如何能在标准注释工具中轻松地与转录组学数据整合。与仅使用转录组学证据相比,这使得注释的蛋白质编码基因数量增加了14%(从3612个增加到4113个)。人工整理进一步使蛋白质编码基因数量增加了9%(达到4493个)。所有这些基因都有RNA测序证据,并且87%被蛋白质组学所证实。本文展示的合轴马拉色菌基因组组装和注释质量很高,仅少数真核生物达到了这种质量,并且构成了未来宿主 - 微生物相互作用研究的重要参考。