Division of Epidemiology and Biostatistics, European Institute of Oncology, Via Ramusio 1, Milan, 20141, Italy.
BMC Med Res Methodol. 2012 Aug 3;12:116. doi: 10.1186/1471-2288-12-116.
For complex diseases like cancer, pooled-analysis of individual data represents a powerful tool to investigate the joint contribution of genetic, phenotypic and environmental factors to the development of a disease. Pooled-analysis of epidemiological studies has many advantages over meta-analysis, and preliminary results may be obtained faster and with lower costs than with prospective consortia.
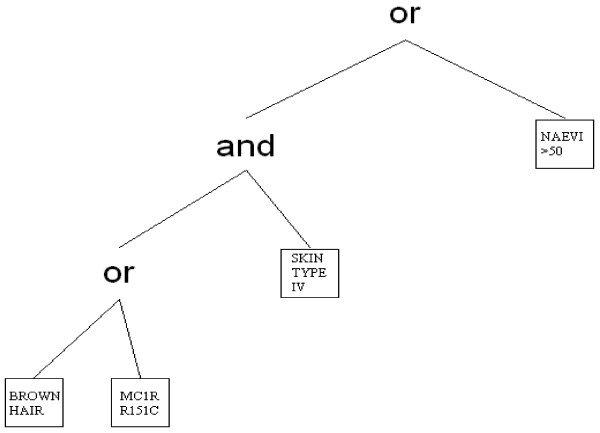
Based on our experience with the study design of the Melanocortin-1 receptor (MC1R) gene, SKin cancer and Phenotypic characteristics (M-SKIP) project, we describe the most important steps in planning and conducting a pooled-analysis of genetic epidemiological studies. We then present the statistical analysis plan that we are going to apply, giving particular attention to methods of analysis recently proposed to account for between-study heterogeneity and to explore the joint contribution of genetic, phenotypic and environmental factors in the development of a disease. Within the M-SKIP project, data on 10,959 skin cancer cases and 14,785 controls from 31 international investigators were checked for quality and recoded for standardization. We first proposed to fit the aggregated data with random-effects logistic regression models. However, for the M-SKIP project, a two-stage analysis will be preferred to overcome the problem regarding the availability of different study covariates. The joint contribution of MC1R variants and phenotypic characteristics to skin cancer development will be studied via logic regression modeling.
Methodological guidelines to correctly design and conduct pooled-analyses are needed to facilitate application of such methods, thus providing a better summary of the actual findings on specific fields.
对于癌症等复杂疾病,个体数据的汇总分析是研究遗传、表型和环境因素共同作用于疾病发生的有力工具。与荟萃分析相比,对流行病学研究进行汇总分析具有许多优势,并且可能比前瞻性联盟更快、成本更低地获得初步结果。
基于我们在黑素皮质素受体 1(MC1R)基因、皮肤癌和表型特征(M-SKIP)项目研究设计方面的经验,我们描述了规划和进行遗传流行病学研究汇总分析的最重要步骤。然后,我们介绍了我们将要应用的统计分析计划,特别关注最近提出的用于解释研究间异质性并探索遗传、表型和环境因素共同作用于疾病发生的分析方法。在 M-SKIP 项目中,来自 31 位国际研究者的 10959 例皮肤癌病例和 14785 例对照者的数据经过质量检查和标准化重新编码。我们最初提议使用随机效应逻辑回归模型拟合汇总数据。然而,对于 M-SKIP 项目,将首选两阶段分析来克服研究协变量可用性的问题。通过逻辑回归建模研究 MC1R 变体和表型特征对皮肤癌发生的共同作用。
需要正确设计和实施汇总分析的方法学指南,以促进这些方法的应用,从而更好地总结特定领域的实际发现。