Research Programme on Biomedical Informatics - GRIB, Universitat Pompeu Fabra - UPF, Parc de Recerca Biomèdica de Barcelona. Dr. Aiguader, 88, E-08003 Barcelona, Spain.
Nucleic Acids Res. 2012 Nov;40(21):e169. doi: 10.1093/nar/gks743. Epub 2012 Aug 16.
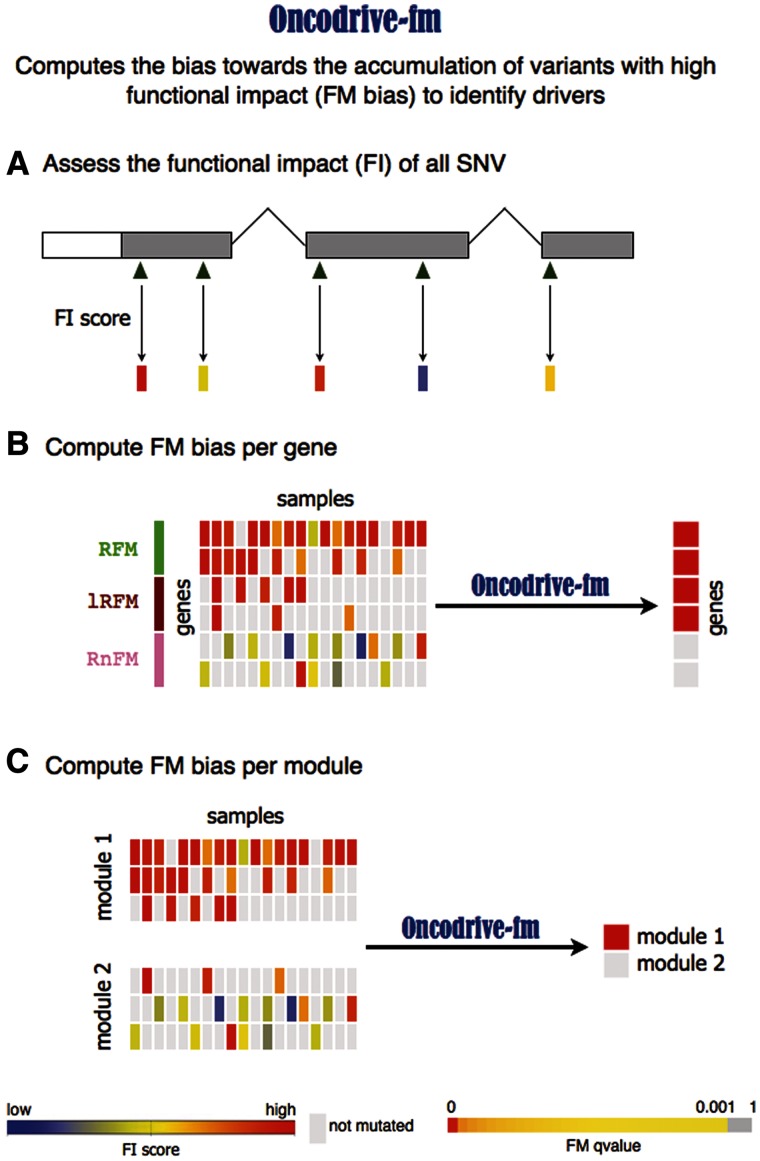
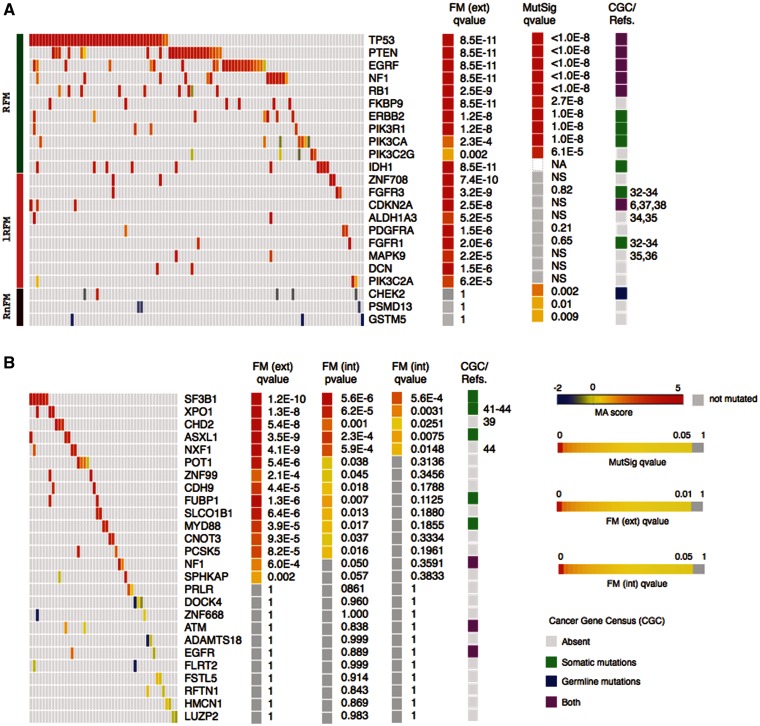
Identifying cancer driver genes and pathways among all somatic mutations detected in a cohort of tumors is a key challenge in cancer genomics. Traditionally, this is done by prioritizing genes according to the recurrence of alterations that they bear. However, this approach has some known limitations, such as the difficulty to correctly estimate the background mutation rate, and the fact that it cannot identify lowly recurrently mutated driver genes. Here we present a novel approach, Oncodrive-fm, to detect candidate cancer drivers which does not rely on recurrence. First, we hypothesized that any bias toward the accumulation of variants with high functional impact observed in a gene or group of genes may be an indication of positive selection and can thus be used to detect candidate driver genes or gene modules. Next, we developed a method to measure this bias (FM bias) and applied it to three datasets of tumor somatic variants. As a proof of concept of our hypothesis we show that most of the highly recurrent and well-known cancer genes exhibit a clear FM bias. Moreover, this novel approach avoids some known limitations of recurrence-based approaches, and can successfully identify lowly recurrent candidate cancer drivers.
在肿瘤队列中检测到的所有体细胞突变中识别癌症驱动基因和途径是癌症基因组学的一个关键挑战。传统上,这是通过根据它们所具有的改变的复发频率来优先考虑基因来完成的。然而,这种方法有一些已知的局限性,例如难以正确估计背景突变率,以及无法识别低频率复发的驱动基因的事实。在这里,我们提出了一种新的方法 Oncodrive-fm,用于检测候选癌症驱动基因,该方法不依赖于复发。首先,我们假设在一个基因或一组基因中观察到的任何偏向于积累具有高功能影响的变体的趋势可能是正选择的迹象,因此可以用于检测候选驱动基因或基因模块。接下来,我们开发了一种测量这种偏差(FM 偏差)的方法,并将其应用于三个肿瘤体细胞变异数据集。作为我们假设的概念验证,我们表明大多数高复发和著名的癌症基因表现出明显的 FM 偏差。此外,这种新方法避免了基于复发的方法的一些已知局限性,并且可以成功识别低频率复发的候选癌症驱动基因。