Institut National de la Recherche Agronomique, UR1052, Unité de Génétique et Amélioration des Fruits et Légumes, Avignon, France.
G3 (Bethesda). 2012 Aug;2(8):853-64. doi: 10.1534/g3.112.002667. Epub 2012 Aug 1.
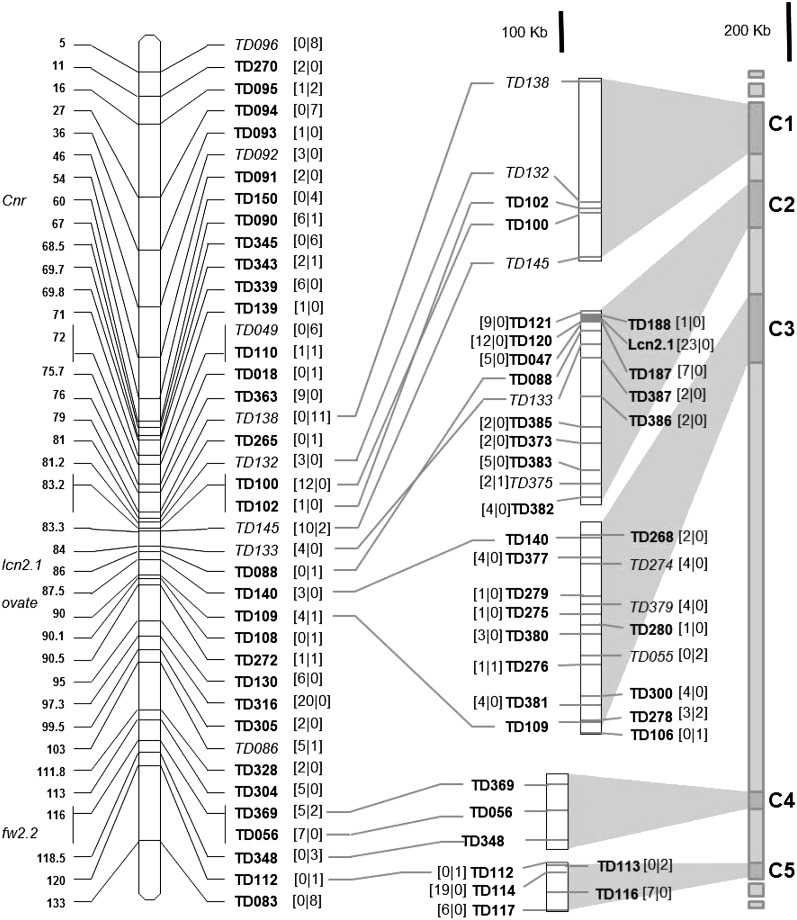
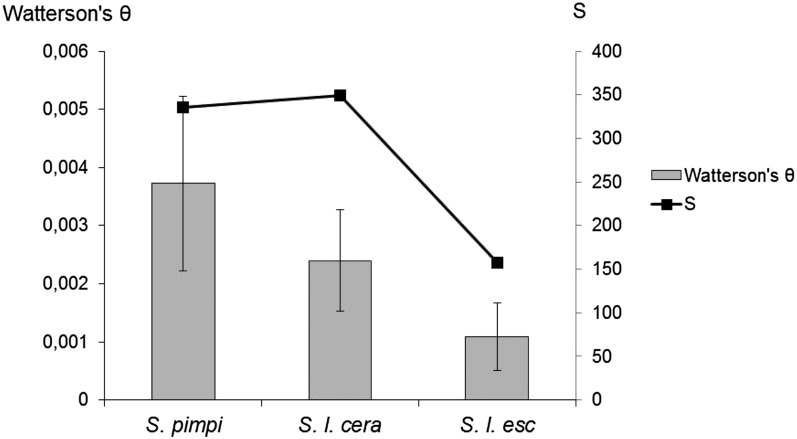
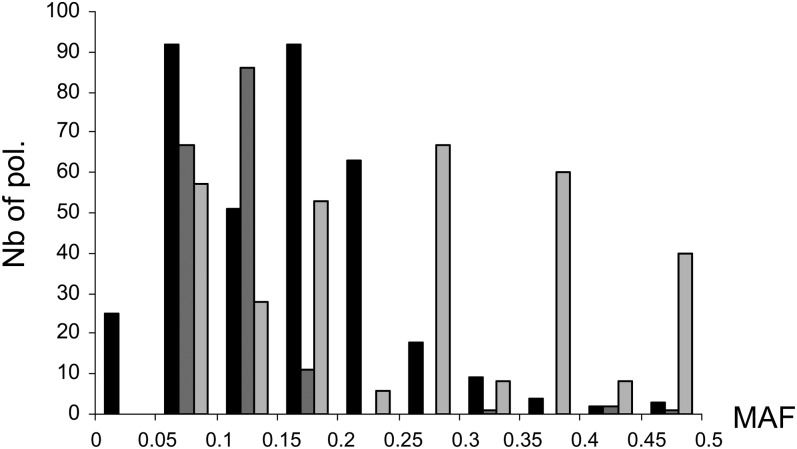
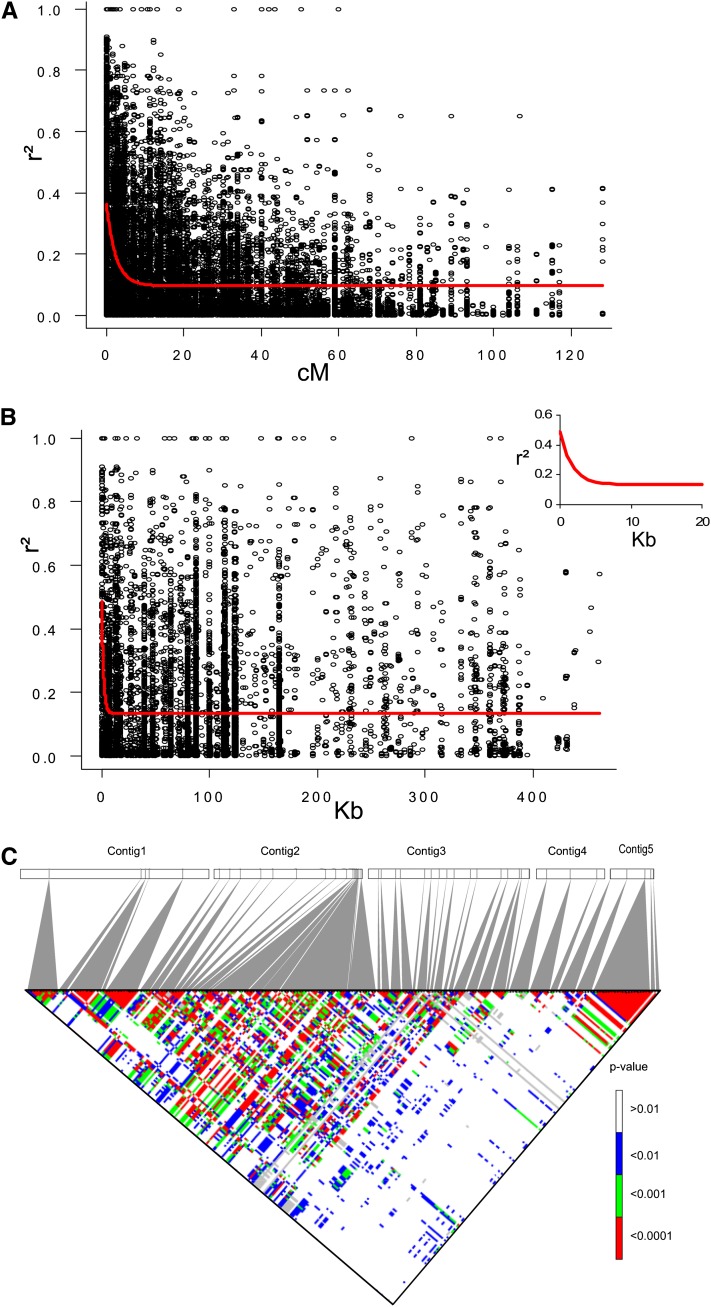
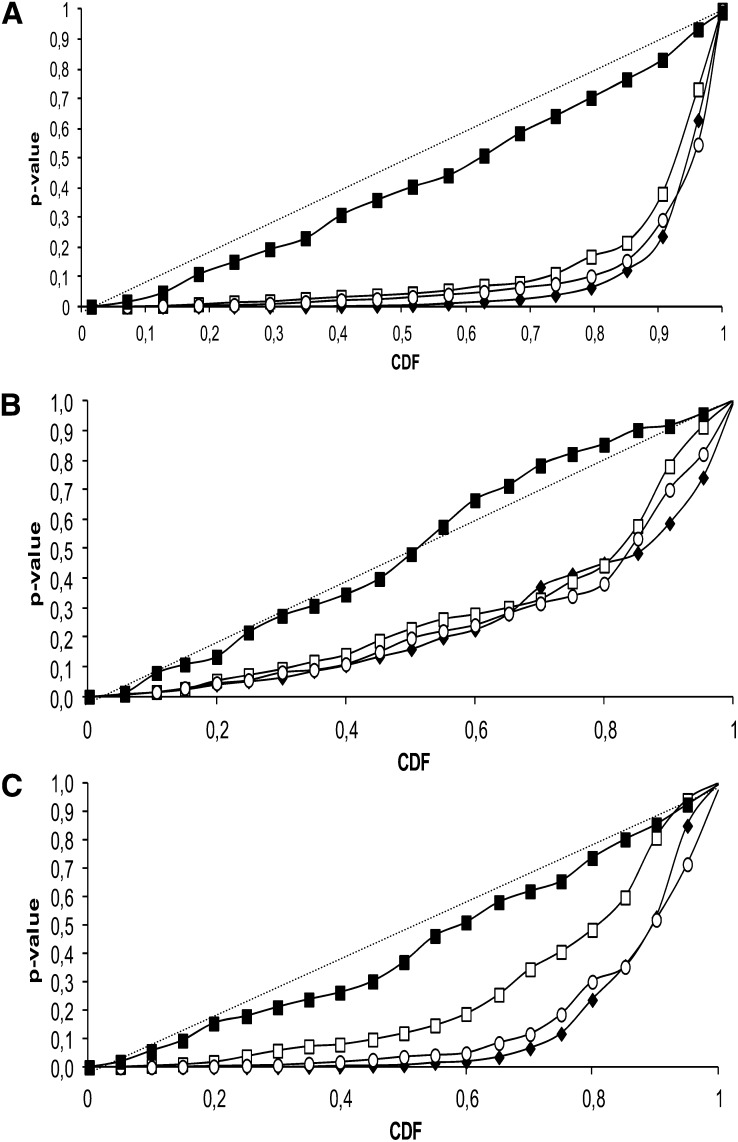
Genome-wide association mapping is an efficient way to identify quantitative trait loci controlling the variation of phenotypes, but the approach suffers severe limitations when one is studying inbred crops like cultivated tomato (Solanum lycopersicum). Such crops exhibit low rates of molecular polymorphism and high linkage disequilibrium, which reduces mapping resolution. The cherry type tomato (S. lycopersicum var. cerasiforme) genome has been described as an admixture between the cultivated tomato and its wild ancestor, S. pimpinellifolium. We have thus taken advantage of the properties of this admixture to improve the resolution of association mapping in tomato. As a proof of concept, we sequenced 81 DNA fragments distributed on chromosome 2 at different distances in a core collection of 90 tomato accessions, including mostly cherry type tomato accessions. The 81 Sequence Tag Sites revealed 352 SNPs and indels. Molecular diversity was greatest for S. pimpinellifolium accessions, intermediate for S. l. cerasiforme accessions, and lowest for the cultivated group. We assessed the structure of molecular polymorphism and the extent of linkage disequilibrium over genetic and physical distances. Linkage disequilibrium decreased under r(2) = 0.3 within 1 cM, and minimal estimated value (r(2) = 0.13) was reached within 20 kb over the physical regions studied. Associations between polymorphisms and fruit weight, locule number, and soluble solid content were detected. Several candidate genes and quantitative trait loci previously identified were validated and new associations detected. This study shows the advantages of using a collection of S. l. cerasiforme accessions to overcome the low resolution of association mapping in tomato.
全基因组关联作图是一种有效的方法,可以识别控制表型变异的数量性状基因座,但在研究像栽培番茄(Solanum lycopersicum)这样的自交作物时,该方法受到严重限制。这些作物表现出低水平的分子多态性和高度的连锁不平衡,从而降低了作图分辨率。樱桃番茄(S. lycopersicum var. cerasiforme)基因组被描述为栽培番茄与其野生祖先 S. pimpinellifolium 的混合物。因此,我们利用这种混合物的特性来提高番茄关联作图的分辨率。作为概念验证,我们对核心番茄品种 90 个品种中的 81 个 DNA 片段进行了测序,这些片段分布在染色体 2 上,距离不同。这 81 个序列标签位点揭示了 352 个 SNPs 和插入缺失。S. pimpinellifolium 品种的分子多样性最大,S. l. cerasiforme 品种的分子多样性居中,而栽培品种的分子多样性最低。我们评估了遗传和物理距离上的分子多态性结构和连锁不平衡程度。r(2) = 0.3 以内的连锁不平衡在 1 cM 内降低,r(2) = 0.13 以内的最小估计值在研究的物理区域内达到 20 kb 以内。检测到多态性与果实重量、心室数和可溶性固形物含量之间的关联。验证了先前鉴定的几个候选基因和数量性状基因座,并检测到新的关联。这项研究表明,利用 S. l. cerasiforme 品种的集合来克服番茄关联作图分辨率低的问题具有优势。