Institut National de la Recherche Agronomique (INRA), Research Unit 1290 BIOGER, Thiverval-Grignon, France.
G3 (Bethesda). 2012 Aug;2(8):891-904. doi: 10.1534/g3.112.002048. Epub 2012 Aug 1.
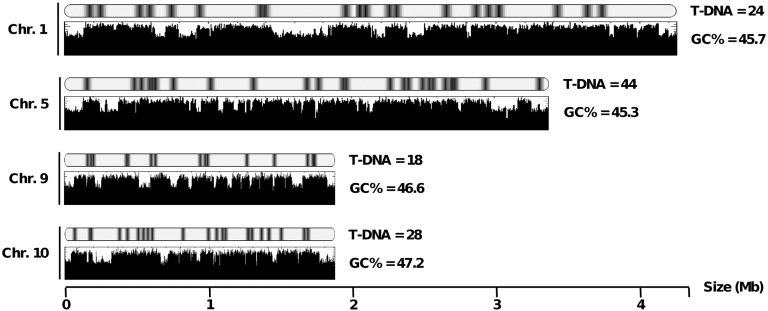
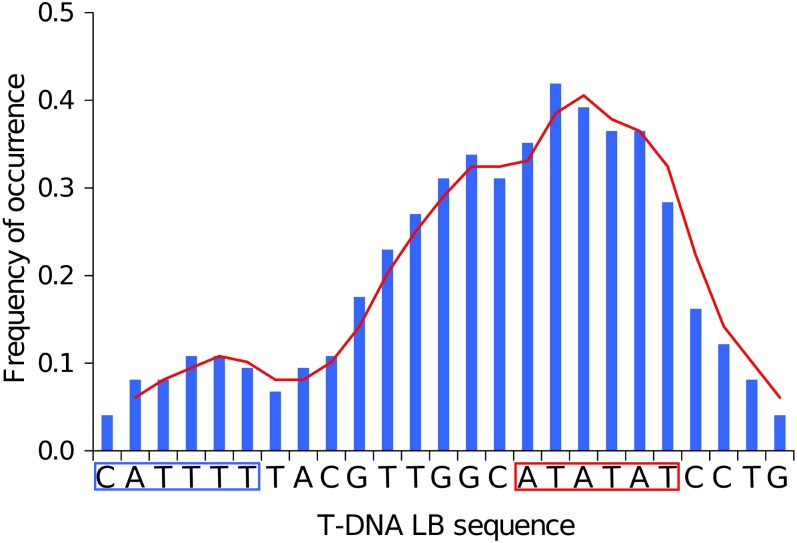
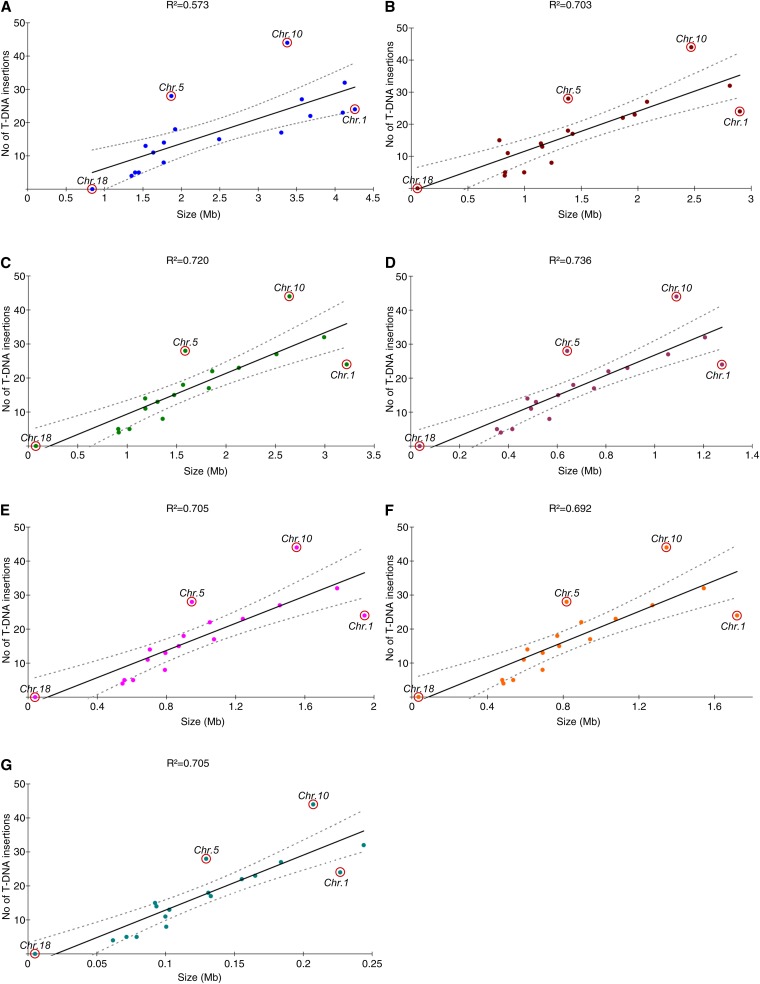
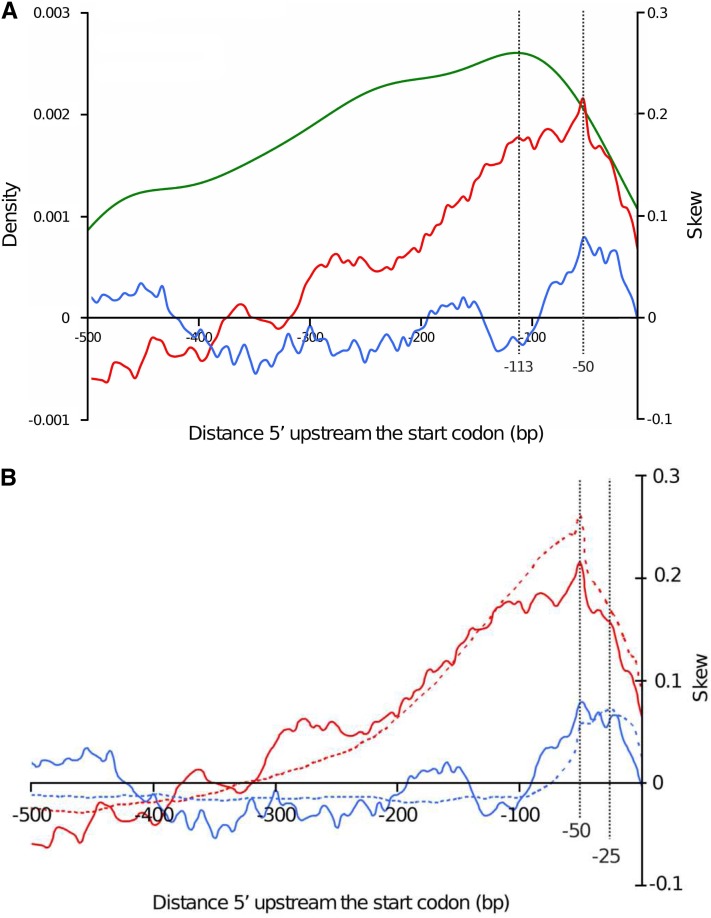
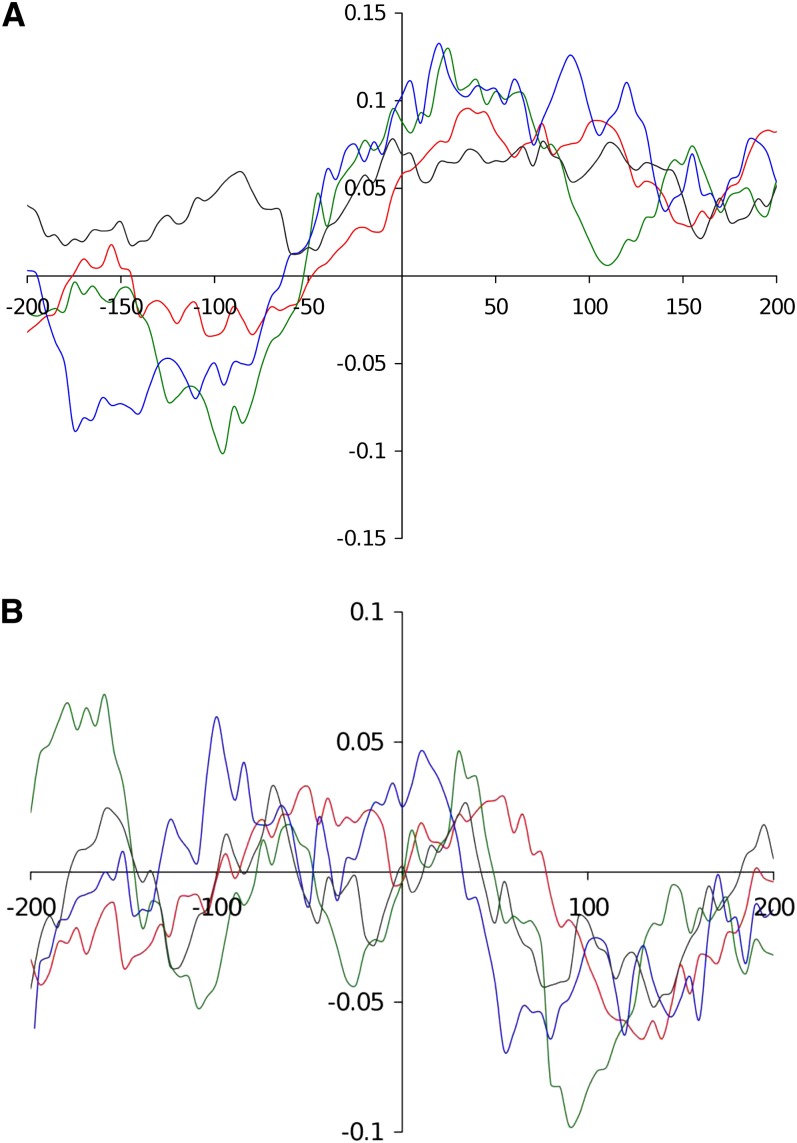
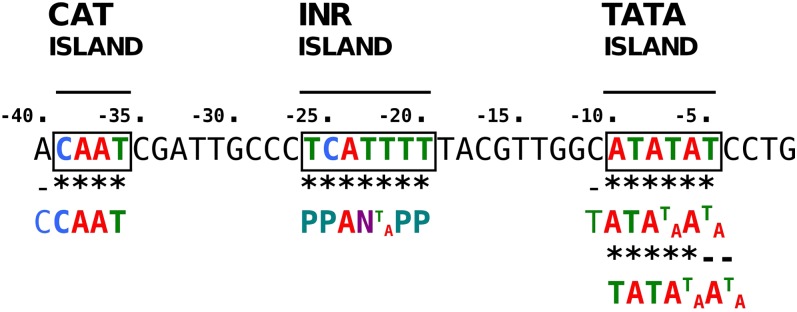
The ever-increasing generation of sequence data is accompanied by unsatisfactory functional annotation, and complex genomes, such as those of plants and filamentous fungi, show a large number of genes with no predicted or known function. For functional annotation of unknown or hypothetical genes, the production of collections of mutants using Agrobacterium tumefaciens-mediated transformation (ATMT) associated with genotyping and phenotyping has gained wide acceptance. ATMT is also widely used to identify pathogenicity determinants in pathogenic fungi. A systematic analysis of T-DNA borders was performed in an ATMT-mutagenized collection of the phytopathogenic fungus Leptosphaeria maculans to evaluate the features of T-DNA integration in its particular transposable element-rich compartmentalized genome. A total of 318 T-DNA tags were recovered and analyzed for biases in chromosome and genic compartments, existence of CG/AT skews at the insertion site, and occurrence of microhomologies between the T-DNA left border (LB) and the target sequence. Functional annotation of targeted genes was done using the Gene Ontology annotation. The T-DNA integration mainly targeted gene-rich, transcriptionally active regions, and it favored biological processes consistent with the physiological status of a germinating spore. T-DNA integration was strongly biased toward regulatory regions, and mainly promoters. Consistent with the T-DNA intranuclear-targeting model, the density of T-DNA insertion correlated with CG skew near the transcription initiation site. The existence of microhomologies between promoter sequences and the T-DNA LB flanking sequence was also consistent with T-DNA integration to host DNA mediated by homologous recombination based on the microhomology-mediated end-joining pathway.
不断增长的序列数据伴随着功能注释不理想的问题,而复杂的基因组,如植物和丝状真菌的基因组,显示出大量具有未知或已知功能的基因。对于未知或假设基因的功能注释,使用根癌农杆菌介导的转化(ATMT)与基因分型和表型相结合生产突变体集合已得到广泛认可。ATMT 也广泛用于鉴定病原真菌中的致病性决定因子。对植物病原真菌长柄壳菌的 ATMT 诱变突变体集合中的 T-DNA 边界进行了系统分析,以评估 T-DNA 整合在其特定转座元件丰富的分隔基因组中的特征。总共回收了 318 个 T-DNA 标签,并分析了它们在染色体和基因区室中的偏倚、插入位点处 CG/AT 倾斜的存在以及 T-DNA 左边界(LB)和靶序列之间微同源的发生情况。使用基因本体论注释对靶向基因进行功能注释。T-DNA 整合主要针对基因丰富、转录活跃的区域,并且有利于与萌发孢子生理状态一致的生物过程。T-DNA 整合强烈偏向于调控区,主要是启动子。与 T-DNA 核内靶向模型一致,T-DNA 插入密度与转录起始位点附近的 CG 倾斜相关。启动子序列与 T-DNA LB 侧翼序列之间微同源的存在也与基于微同源介导的末端连接途径的同源重组介导的 T-DNA 整合到宿主 DNA 一致。