Federico Maria, Leoncini Mauro, Montangero Manuela, Valente Paolo
Dipartimento di Scienze Fisiche, Informatiche e Matematiche, Università di Modena e Reggio Emilia, 41125 Modena, Via Campi 213/b, Italy.
Algorithms Mol Biol. 2012 Aug 21;7(1):20. doi: 10.1186/1748-7188-7-20.
The notion of DNA motif is a mathematical abstraction used to model regions of the DNA (known as Transcription Factor Binding Sites, or TFBSs) that are bound by a given Transcription Factor to regulate gene expression or repression. In turn, DNA structured motifs are a mathematical counterpart that models sets of TFBSs that work in concert in the gene regulations processes of higher eukaryotic organisms. Typically, a structured motif is composed of an ordered set of isolated (or simple) motifs, separated by a variable, but somewhat constrained number of "irrelevant" base-pairs. Discovering structured motifs in a set of DNA sequences is a computationally hard problem that has been addressed by a number of authors using either a direct approach, or via the preliminary identification and successive combination of simple motifs.
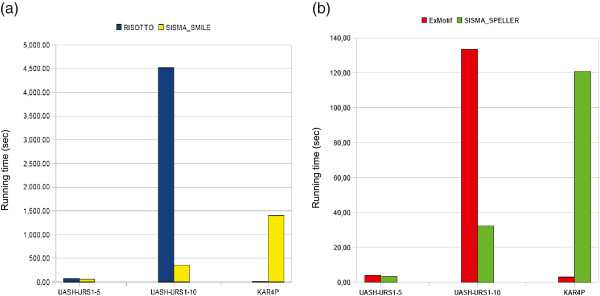
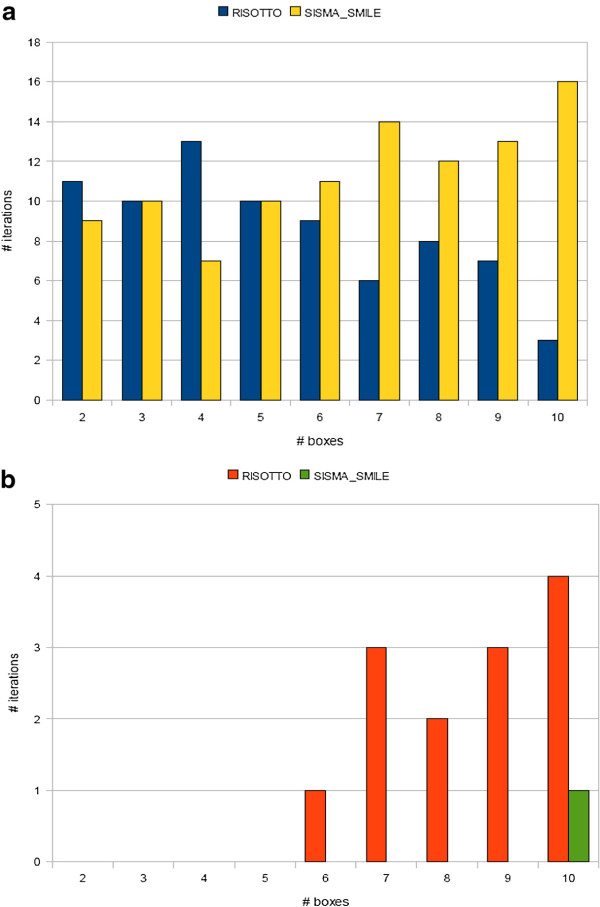
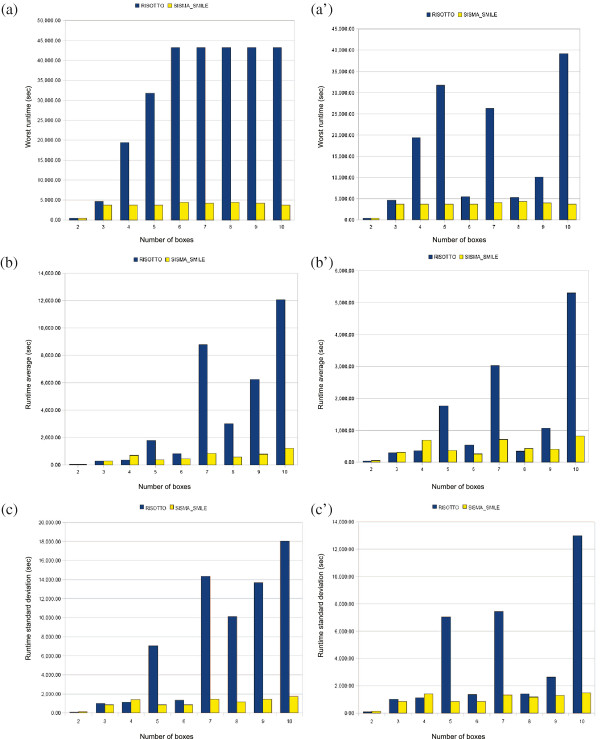
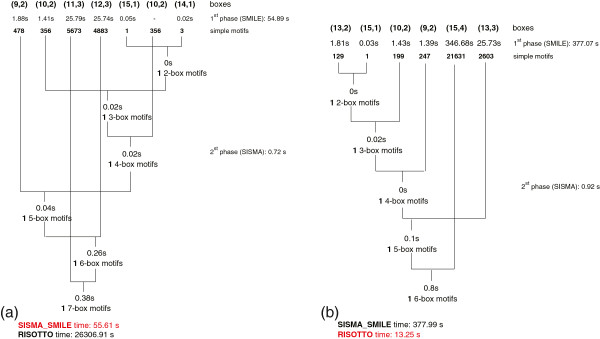
We describe a computational tool, named SISMA, for the de-novo discovery of structured motifs in a set of DNA sequences. SISMA is an exact, enumerative algorithm, meaning that it finds all the motifs conforming to the specifications. It does so in two stages: first it discovers all the possible component simple motifs, then combines them in a way that respects the given constraints. We developed SISMA mainly with the aim of understanding the potential benefits of such a 2-stage approach w.r.t. direct methods. In fact, no 2-stage software was available for the general problem of structured motif discovery, but only a few tools that solved restricted versions of the problem. We evaluated SISMA against other published tools on a comprehensive benchmark made of both synthetic and real biological datasets. In a significant number of cases, SISMA outperformed the competitors, exhibiting a good performance also in most of the cases in which it was inferior.
A reflection on the results obtained lead us to conclude that a 2-stage approach can be implemented with many advantages over direct approaches. Some of these have to do with greater modularity, ease of parallelization, and the possibility to perform adaptive searches of structured motifs. As another consideration, we noted that most hard instances for SISMA were easy to detect in advance. In these cases one may initially opt for a direct method; or, as a viable alternative in most laboratories, one could run both direct and 2-stage tools in parallel, halting the computations when the first halts.
DNA基序的概念是一种数学抽象,用于对DNA区域(称为转录因子结合位点,或TFBS)进行建模,这些区域被特定的转录因子结合以调节基因表达或抑制。反过来,DNA结构化基序是一种数学对应物,用于对在高等真核生物基因调控过程中协同作用的TFBS集合进行建模。通常,一个结构化基序由一组有序的孤立(或简单)基序组成,它们之间由数量可变但有一定限制的“无关”碱基对分隔。在一组DNA序列中发现结构化基序是一个计算难题,许多作者使用直接方法或通过简单基序的初步识别和连续组合来解决这个问题。
我们描述了一种名为SISMA的计算工具,用于在一组DNA序列中从头发现结构化基序。SISMA是一种精确的枚举算法,这意味着它会找到所有符合规范的基序。它分两个阶段进行:首先它发现所有可能的组成简单基序,然后以符合给定约束的方式将它们组合起来。我们开发SISMA主要是为了了解这种两阶段方法相对于直接方法的潜在优势。事实上,对于结构化基序发现的一般问题,没有可用的两阶段软件,只有少数工具解决了该问题的受限版本。我们在由合成和真实生物数据集组成的综合基准上,将SISMA与其他已发表的工具进行了评估。在大量情况下,SISMA优于竞争对手,在大多数不如竞争对手表现的情况下也表现出良好的性能。
对所获得结果的思考使我们得出结论,两阶段方法相对于直接方法具有许多优势。其中一些优势与更高的模块化、易于并行化以及对结构化基序进行自适应搜索的可能性有关。作为另一个考虑因素,我们注意到SISMA的大多数困难实例很容易预先检测到。在这些情况下,人们可以最初选择直接方法;或者,作为大多数实验室可行的替代方法,可以并行运行直接工具和两阶段工具,当第一个工具停止时停止计算。