Computer Science Department, ETH Zürich, Universitätsstrasse 6, CH-8092 Zürich, Switzerland.
Nucleic Acids Res. 2012 Nov 1;40(20):10005-17. doi: 10.1093/nar/gks726. Epub 2012 Aug 25.
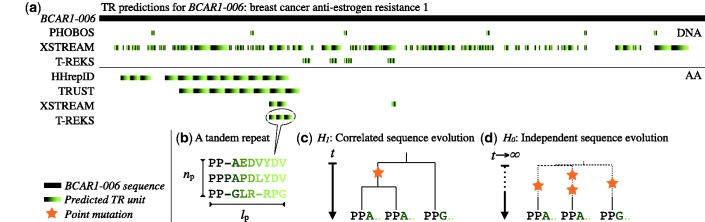
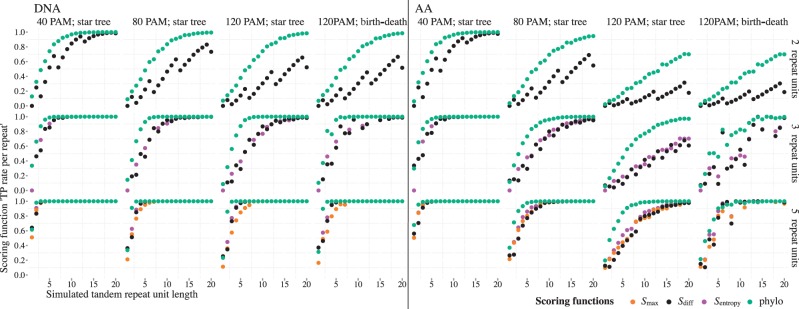
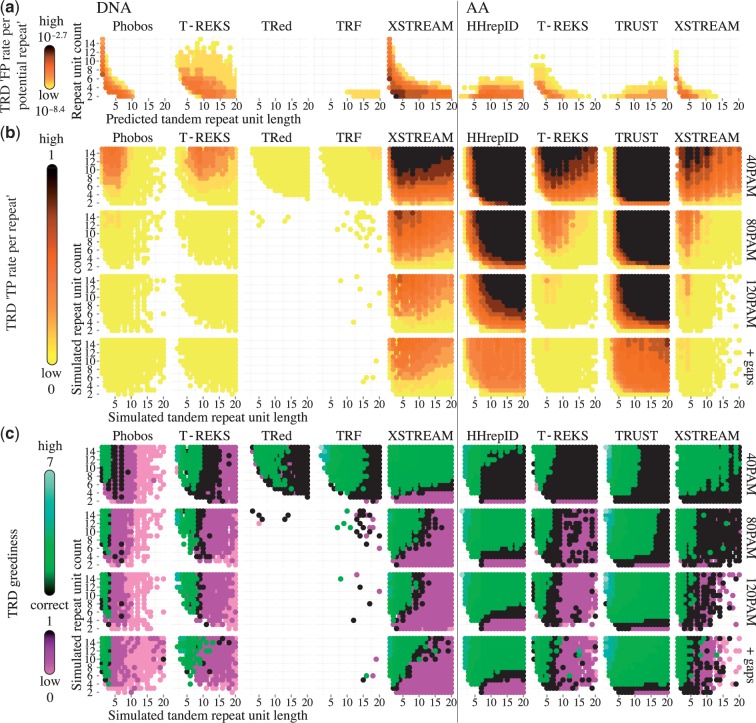
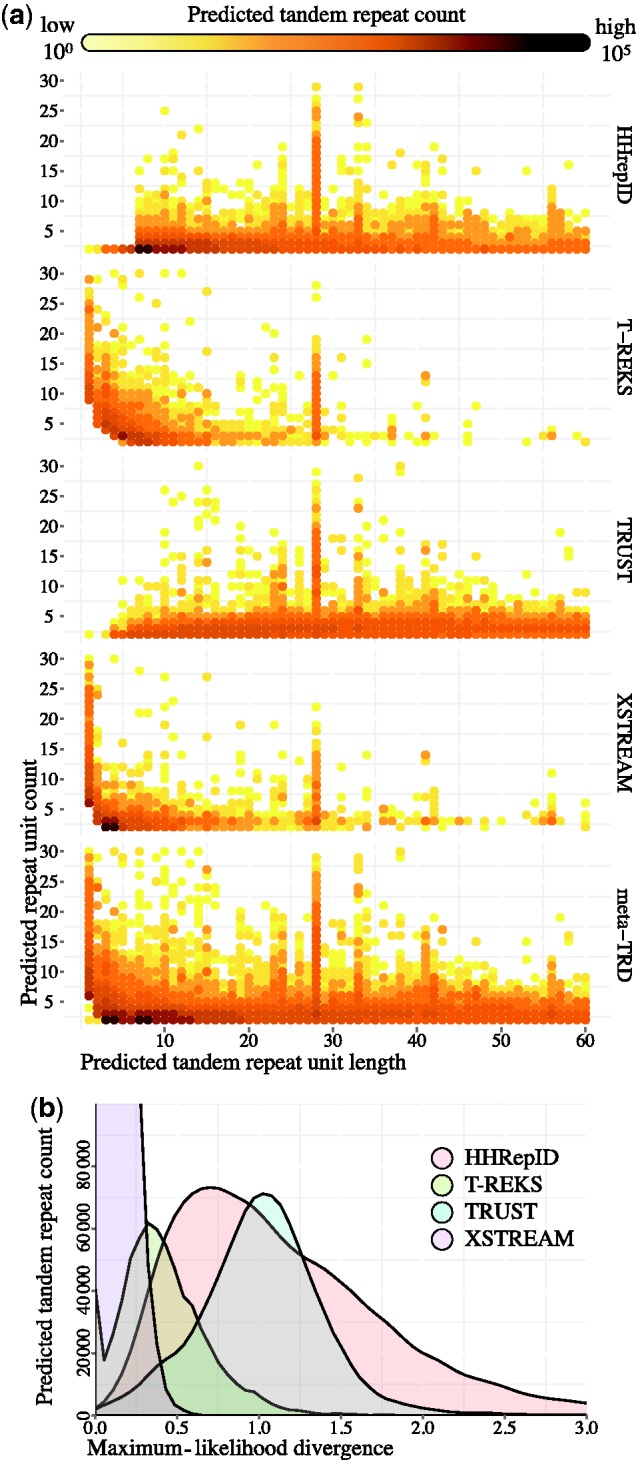
Tandem repeats (TRs) represent one of the most prevalent features of genomic sequences. Due to their abundance and functional significance, a plethora of detection tools has been devised over the last two decades. Despite the longstanding interest, TR detection is still not resolved. Our large-scale tests reveal that current detectors produce different, often nonoverlapping inferences, reflecting characteristics of the underlying algorithms rather than the true distribution of TRs in genomic data. Our simulations show that the power of detecting TRs depends on the degree of their divergence, and repeat characteristics such as the length of the minimal repeat unit and their number in tandem. To reconcile the diverse predictions of current algorithms, we propose and evaluate several statistical criteria for measuring the quality of predicted repeat units. In particular, we propose a model-based phylogenetic classifier, entailing a maximum-likelihood estimation of the repeat divergence. Applied in conjunction with the state of the art detectors, our statistical classification scheme for inferred repeats allows to filter out false-positive predictions. Since different algorithms appear to specialize at predicting TRs with certain properties, we advise applying multiple detectors with subsequent filtering to obtain the most complete set of genuine repeats.
串联重复序列 (TRs) 是基因组序列中最常见的特征之一。由于其丰富性和功能意义,在过去的二十年中,已经设计出了大量的检测工具。尽管人们对此一直很感兴趣,但 TR 的检测仍然没有得到解决。我们的大规模测试表明,当前的检测器产生了不同的、通常不重叠的推断,这反映了底层算法的特征,而不是 TR 在基因组数据中的真实分布。我们的模拟表明,检测 TR 的能力取决于它们的分歧程度,以及重复的特征,如最小重复单元的长度及其串联的数量。为了协调当前算法的不同预测,我们提出并评估了几种用于测量预测重复单元质量的统计标准。特别是,我们提出了一种基于模型的系统发育分类器,涉及重复分歧的最大似然估计。与最先进的检测器结合使用,我们对推断重复的统计分类方案可以过滤掉假阳性预测。由于不同的算法似乎专门用于预测具有某些特性的 TR,因此我们建议使用多个检测器进行后续过滤,以获得最完整的真实重复集。