Möller Gabriele, Temml Veronika, Cala Peralta Antonio, Gruet Océane, Richomme Pascal, Séraphin Denis, Viault Guillaume, Kraus Luisa, Huber-Cantonati Petra, Schopfhauser Elisabeth, Pachmayr Johanna, Tokarz Janina, Schuster Daniela, Helesbeux Jean-Jacques, Dyar Kenneth Allen
Institute for Diabetes and Cancer, Helmholtz Center Munich, German Research Center for Environmental Health, 85764 Neuherberg, Germany.
Department of Pharmaceutical and Medicinal Chemistry, Institute of Pharmacy, Paracelsus Medical University Salzburg, 5020 Salzburg, Austria.
Metabolites. 2022 Jan 21;12(2):99. doi: 10.3390/metabo12020099.
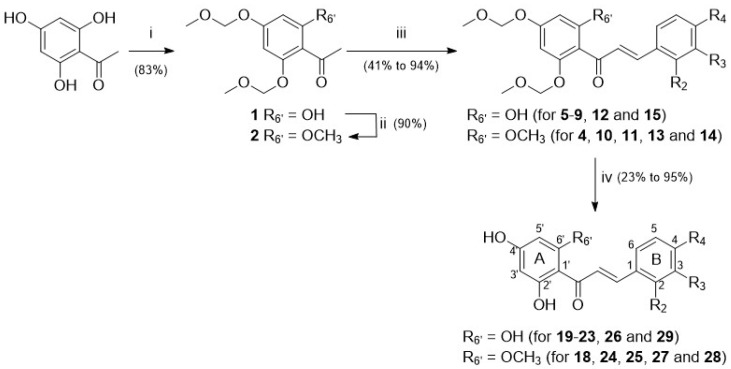
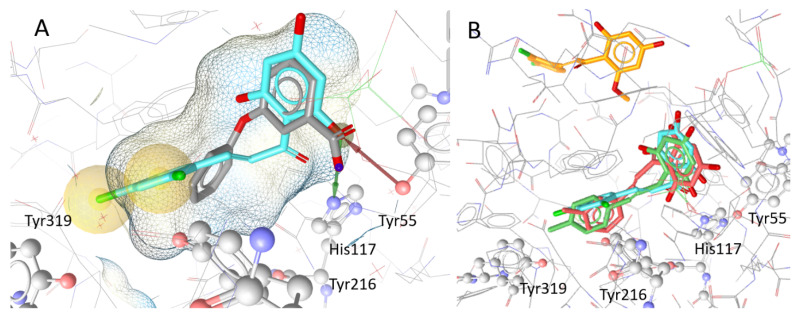
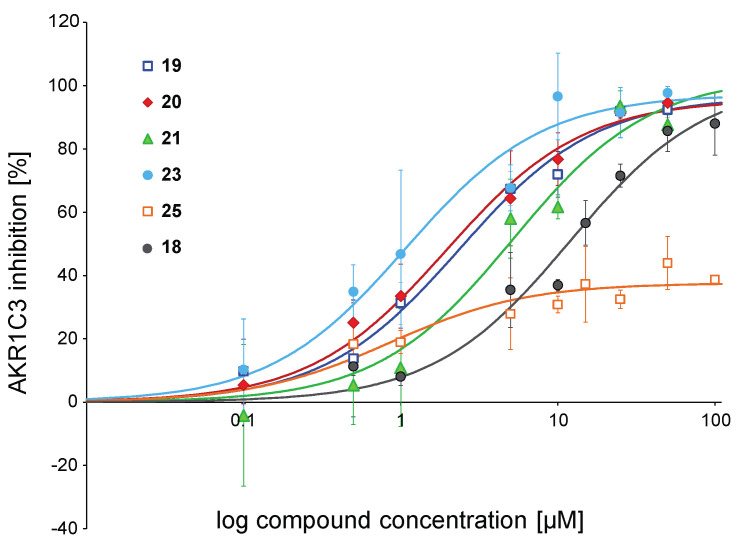
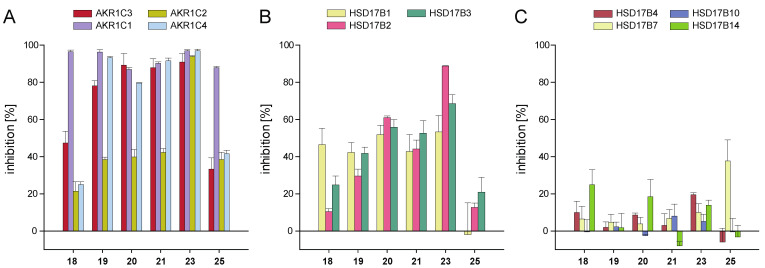
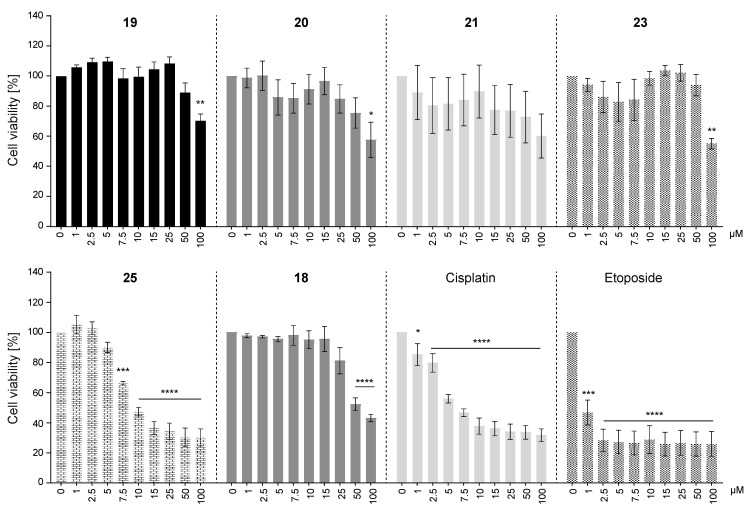
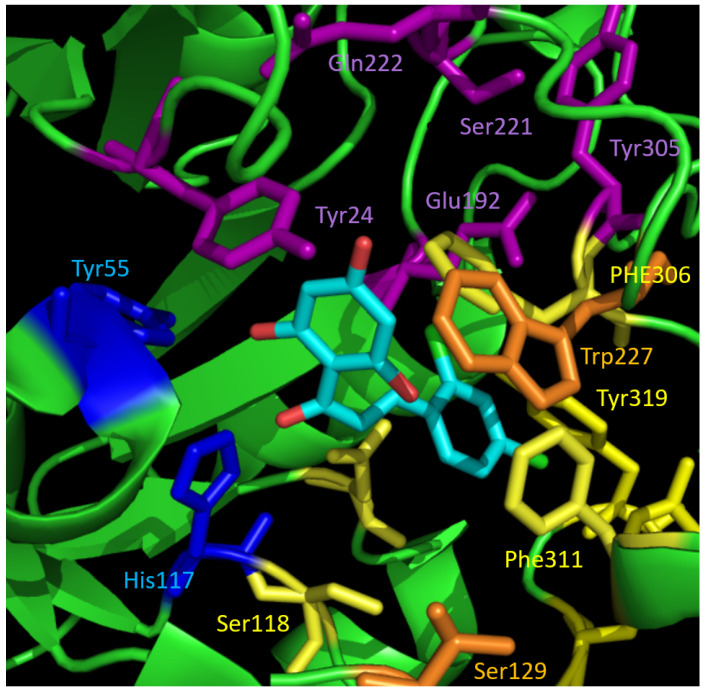
Naturally occurring substances are valuable resources for drug development. In this respect, chalcones are known to be antiproliferative agents against prostate cancer cell lines through various mechanisms or targets. Based on the literature and preliminary results, we aimed to study and optimise the efficiency of a series of chalcones to inhibit androgen-converting AKR1C3, known to promote prostate cancer. A total of 12 chalcones with different substitution patterns were synthesised. Structure-activity relationships associated with these modifications on AKR1C3 inhibition were analysed by performing enzymatic assays and docking simulations. In addition, the selectivity and cytotoxicity of the compounds were assessed. In enzymatic assays, C-6' hydroxylated derivatives were more active than C-6' methoxylated derivatives. In contrast, C-4 methylation increased activity over C-4 hydroxylation. Docking results supported these findings with the most active compounds fitting nicely in the binding site and exhibiting strong interactions with key amino acid residues. The most effective inhibitors were not cytotoxic for HEK293T cells and selective for 17β-hydroxysteroid dehydrogenases not primarily involved in steroid hormone metabolism. Nevertheless, they inhibited several enzymes of the steroid metabolism pathways. Favourable substitutions that enhanced AKR1C3 inhibition of chalcones were identified. This study paves the way to further develop compounds from this series or related flavonoids with improved inhibitory activity against AKR1C3.
天然存在的物质是药物开发的宝贵资源。在这方面,已知查尔酮通过多种机制或靶点对前列腺癌细胞系具有抗增殖作用。基于文献和初步结果,我们旨在研究并优化一系列查尔酮抑制雄激素转化酶AKR1C3的效率,已知该酶可促进前列腺癌。总共合成了12种具有不同取代模式的查尔酮。通过进行酶活性测定和对接模拟分析了与这些修饰对AKR1C3抑制作用相关的构效关系。此外,还评估了化合物的选择性和细胞毒性。在酶活性测定中,C-6'羟基化衍生物比C-6'甲氧基化衍生物更具活性。相反,C-4甲基化比C-4羟基化增加了活性。对接结果支持了这些发现,最具活性的化合物能很好地契合结合位点并与关键氨基酸残基表现出强烈相互作用。最有效的抑制剂对HEK293T细胞无细胞毒性,且对主要不参与类固醇激素代谢的17β-羟基类固醇脱氢酶具有选择性。然而,它们抑制了类固醇代谢途径中的几种酶。确定了增强查尔酮对AKR1C3抑制作用的有利取代基。这项研究为进一步开发该系列化合物或具有改进的对AKR1C3抑制活性的相关黄酮类化合物铺平了道路。