Department of Botany and Plant Sciences, University of California Riverside, Riverside, California, USA.
PLoS One. 2012;7(8):e44173. doi: 10.1371/journal.pone.0044173. Epub 2012 Aug 28.
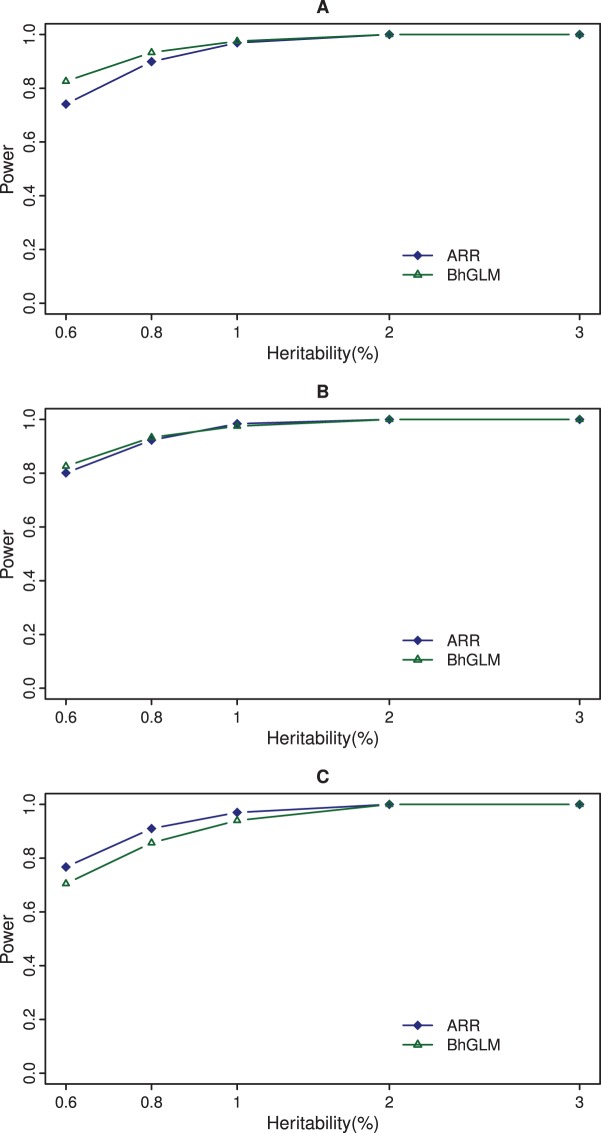
It is widely believed that both common and rare variants contribute to the risks of common diseases or complex traits and the cumulative effects of multiple rare variants can explain a significant proportion of trait variances. Advances in high-throughput DNA sequencing technologies allow us to genotype rare causal variants and investigate the effects of such rare variants on complex traits. We developed an adaptive ridge regression method to analyze the collective effects of multiple variants in the same gene or the same functional unit. Our model focuses on continuous trait and incorporates covariate factors to remove potential confounding effects. The proposed method estimates and tests multiple rare variants collectively but does not depend on the assumption of same direction of each rare variant effect. Compared with the Bayesian hierarchical generalized linear model approach, the state-of-the-art method of rare variant detection, the proposed new method is easy to implement, yet it has higher statistical power. Application of the new method is demonstrated using the well-known data from the Dallas Heart Study.
人们普遍认为,常见变异和罕见变异都可能导致常见疾病或复杂特征的发生,并且多个罕见变异的累积效应可以解释特征变异的很大一部分原因。高通量 DNA 测序技术的进步使我们能够对罕见的因果变异进行基因分型,并研究这些罕见变异对复杂特征的影响。我们开发了一种自适应脊回归方法来分析同一基因或同一功能单元中多个变异的综合效应。我们的模型专注于连续特征,并纳入协变量因素以消除潜在的混杂效应。该方法集中估计和检验多个罕见变异,但不依赖于每个罕见变异效应方向相同的假设。与贝叶斯层次广义线性模型方法相比,该方法是一种用于罕见变异检测的最新方法,所提出的新方法易于实现,但具有更高的统计功效。使用达拉斯心脏研究中著名的数据来演示新方法的应用。