Complex Carbohydrate Research Center, University of Georgia, Athens, GA 30602, USA.
Mol Biol Cell. 2012 Nov;23(21):4175-87. doi: 10.1091/mbc.E12-05-0411. Epub 2012 Sep 5.
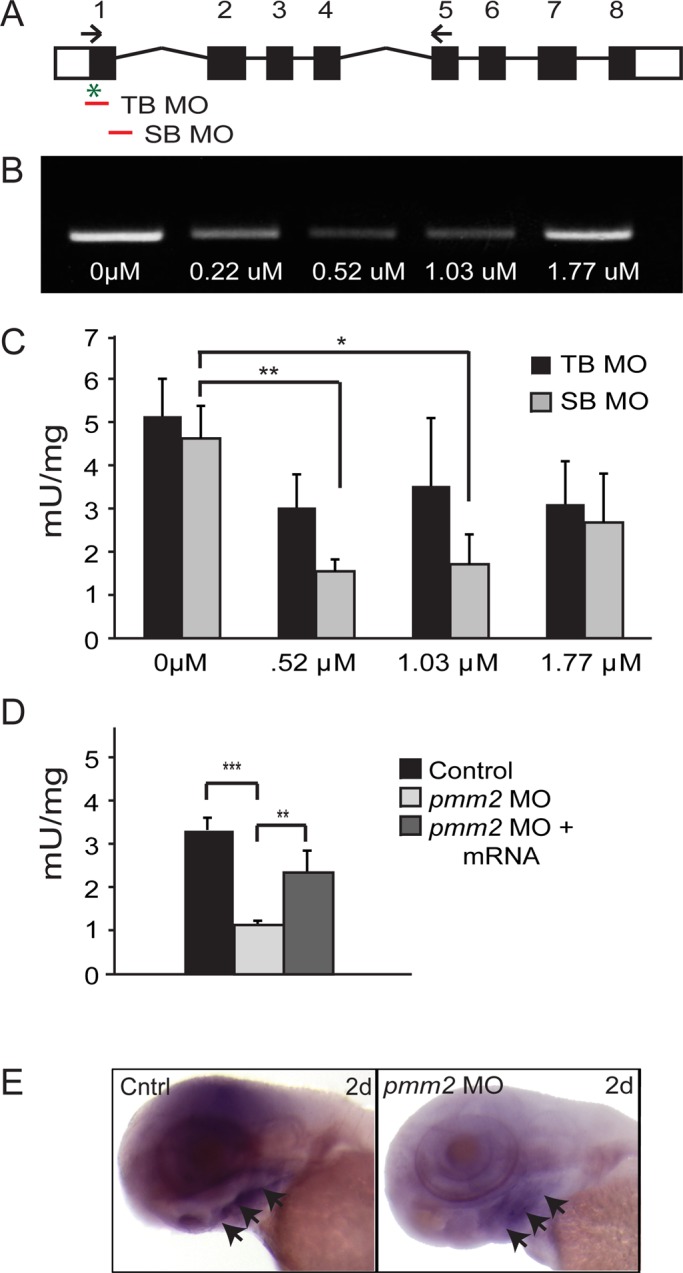
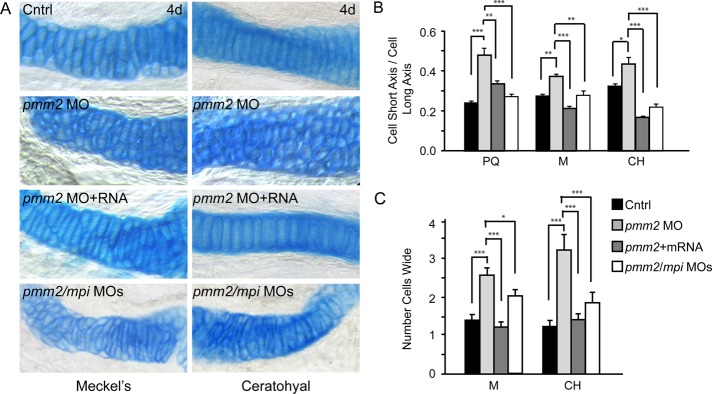
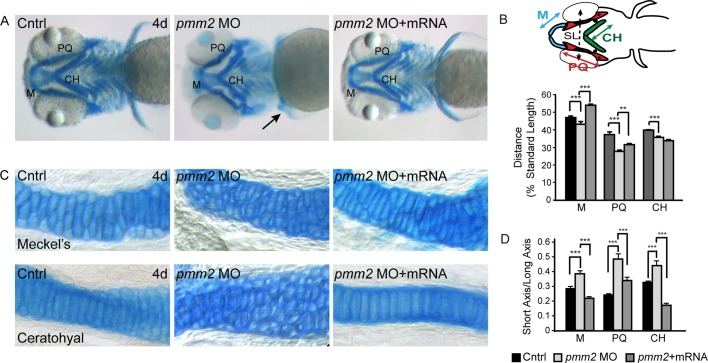
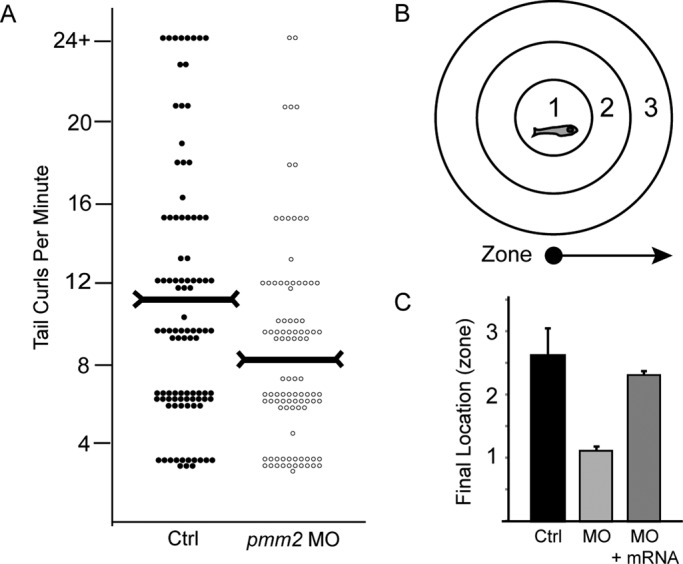
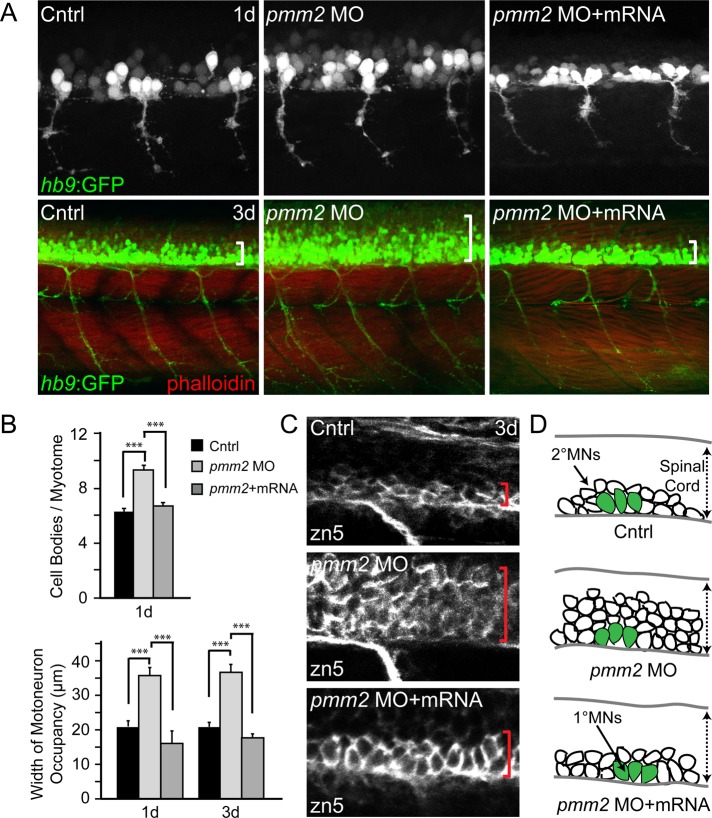
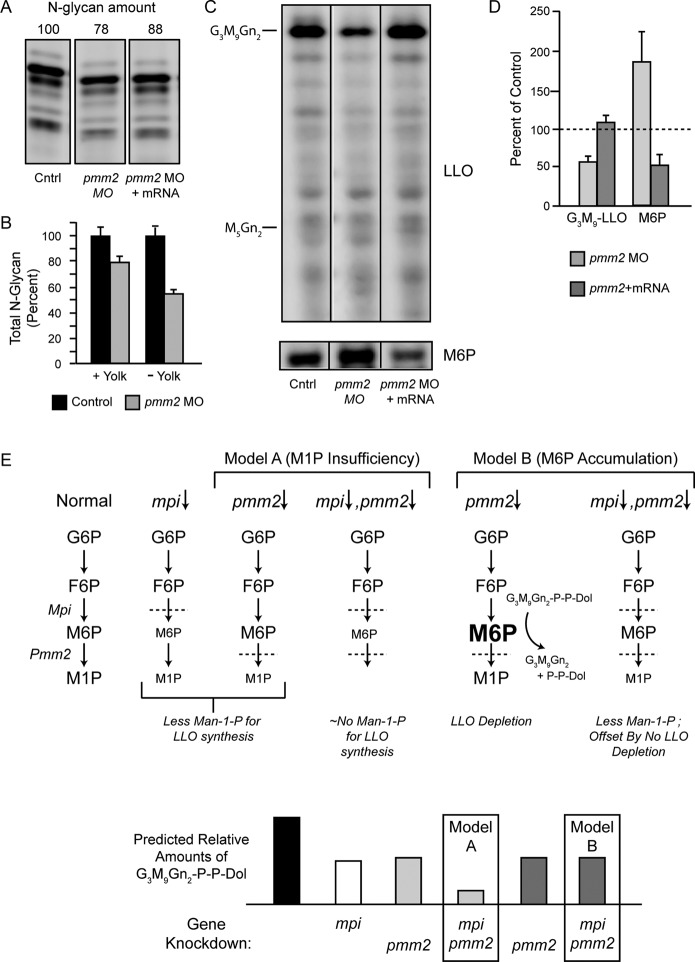
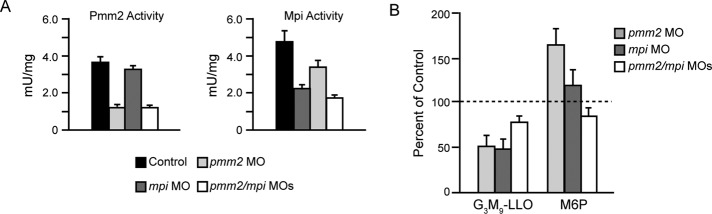
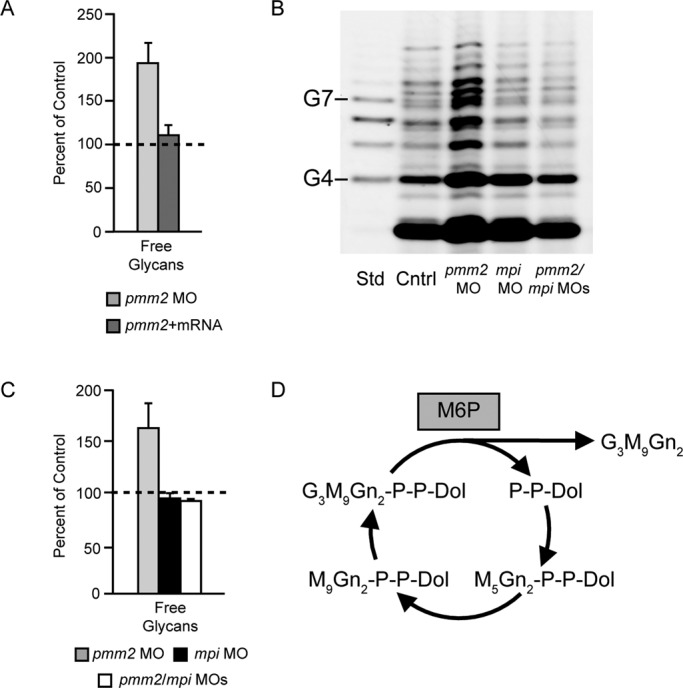
Congenital disorder of glycosylation (PMM2-CDG) results from mutations in pmm2, which encodes the phosphomannomutase (Pmm) that converts mannose-6-phosphate (M6P) to mannose-1-phosphate (M1P). Patients have wide-spectrum clinical abnormalities associated with impaired protein N-glycosylation. Although it has been widely proposed that Pmm2 deficiency depletes M1P, a precursor of GDP-mannose, and consequently suppresses lipid-linked oligosaccharide (LLO) levels needed for N-glycosylation, these deficiencies have not been demonstrated in patients or any animal model. Here we report a morpholino-based PMM2-CDG model in zebrafish. Morphant embryos had developmental abnormalities consistent with PMM2-CDG patients, including craniofacial defects and impaired motility associated with altered motor neurogenesis within the spinal cord. Significantly, global N-linked glycosylation and LLO levels were reduced in pmm2 morphants. Although M1P and GDP-mannose were below reliable detection/quantification limits, Pmm2 depletion unexpectedly caused accumulation of M6P, shown earlier to promote LLO cleavage in vitro. In pmm2 morphants, the free glycan by-products of LLO cleavage increased nearly twofold. Suppression of the M6P-synthesizing enzyme mannose phosphate isomerase within the pmm2 background normalized M6P levels and certain aspects of the craniofacial phenotype and abrogated pmm2-dependent LLO cleavage. In summary, we report the first zebrafish model of PMM2-CDG and uncover novel cellular insights not possible with other systems, including an M6P accumulation mechanism for underglycosylation.
先天性糖基化障碍(PMM2-CDG)是由于编码磷酸甘露糖变位酶(Pmm)的 pmm2 基因突变引起的,该酶将甘露糖-6-磷酸(M6P)转化为甘露糖-1-磷酸(M1P)。患者有广泛的临床异常,与蛋白质 N-糖基化受损有关。尽管广泛提出 Pmm2 缺乏会耗尽 GDP-甘露糖的前体 M1P,从而抑制 N-糖基化所需的脂联寡糖(LLO)水平,但这些缺乏在患者或任何动物模型中都没有得到证明。在这里,我们报告了一种基于 morpholino 的斑马鱼 PMM2-CDG 模型。 Morphant 胚胎具有与 PMM2-CDG 患者一致的发育异常,包括颅面缺陷和运动障碍,这与脊髓内运动神经元发生改变有关。重要的是,pmm2 突变体中的全局 N-连接糖基化和 LLO 水平降低。尽管 M1P 和 GDP-甘露糖低于可靠的检测/定量限,但 Pmm2 耗竭出人意料地导致 M6P 积累,先前的研究表明 M6P 可促进体外 LLO 切割。在 pmm2 突变体中,LLO 切割的游离糖副产物增加了近两倍。在 pmm2 背景下抑制 M6P 合成酶磷酸甘露糖异构酶可使 M6P 水平正常化,并可使某些颅面表型和 pmm2 依赖性 LLO 切割正常化。总之,我们报告了第一个 PMM2-CDG 斑马鱼模型,并揭示了其他系统不可能获得的新的细胞见解,包括一种用于低糖基化的 M6P 积累机制。