Center for Clinical and Translational Science, The Rockefeller University, New York, New York, United States of America.
PLoS One. 2012;7(9):e44274. doi: 10.1371/journal.pone.0044274. Epub 2012 Sep 5.
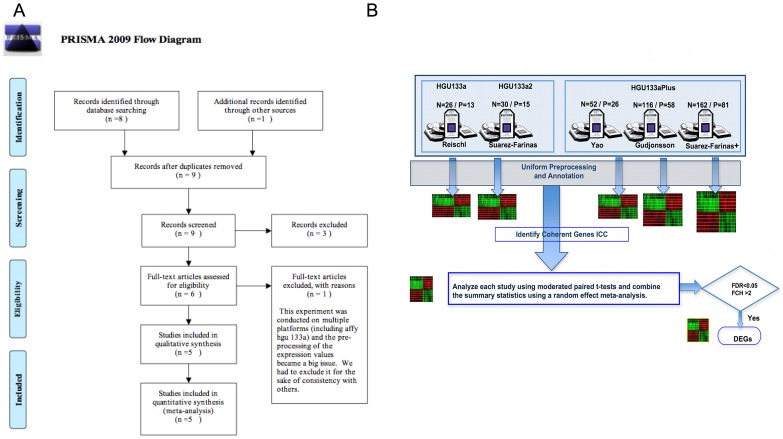
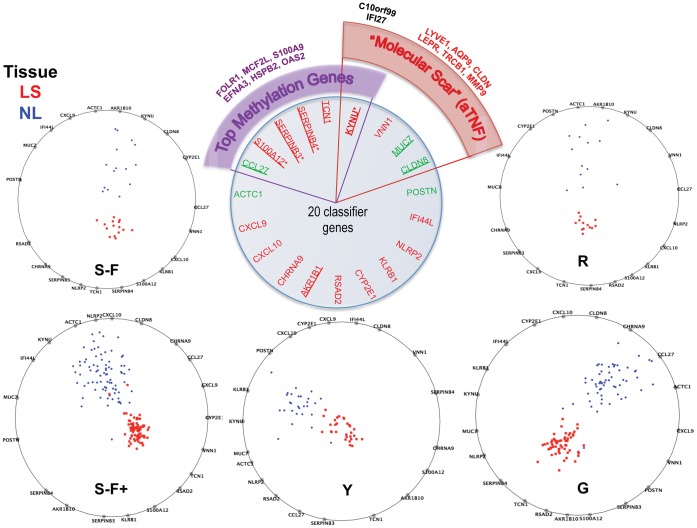
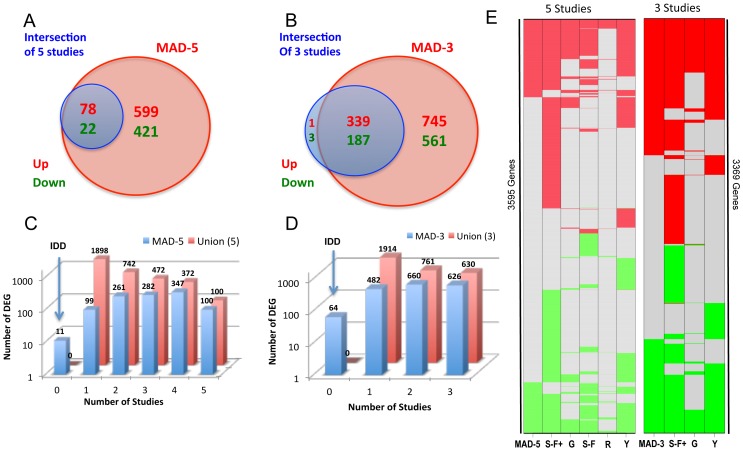
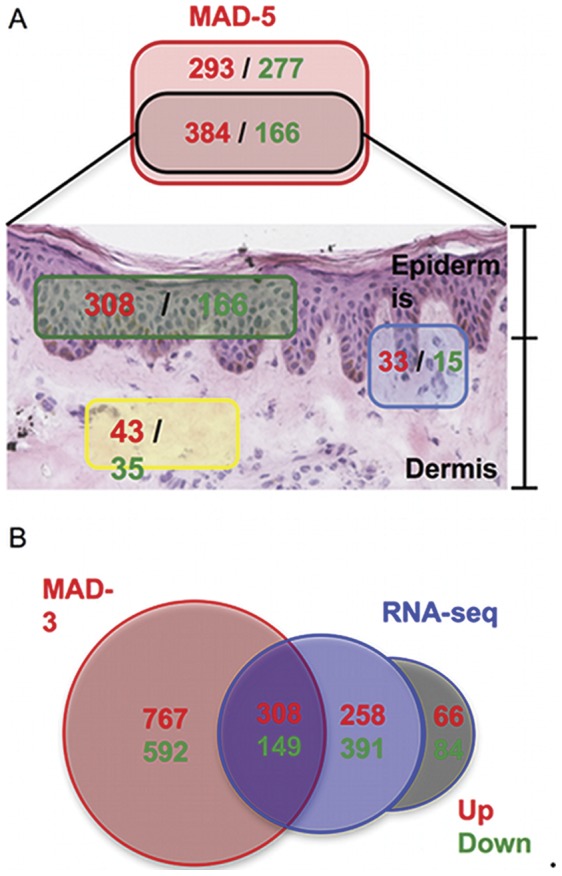
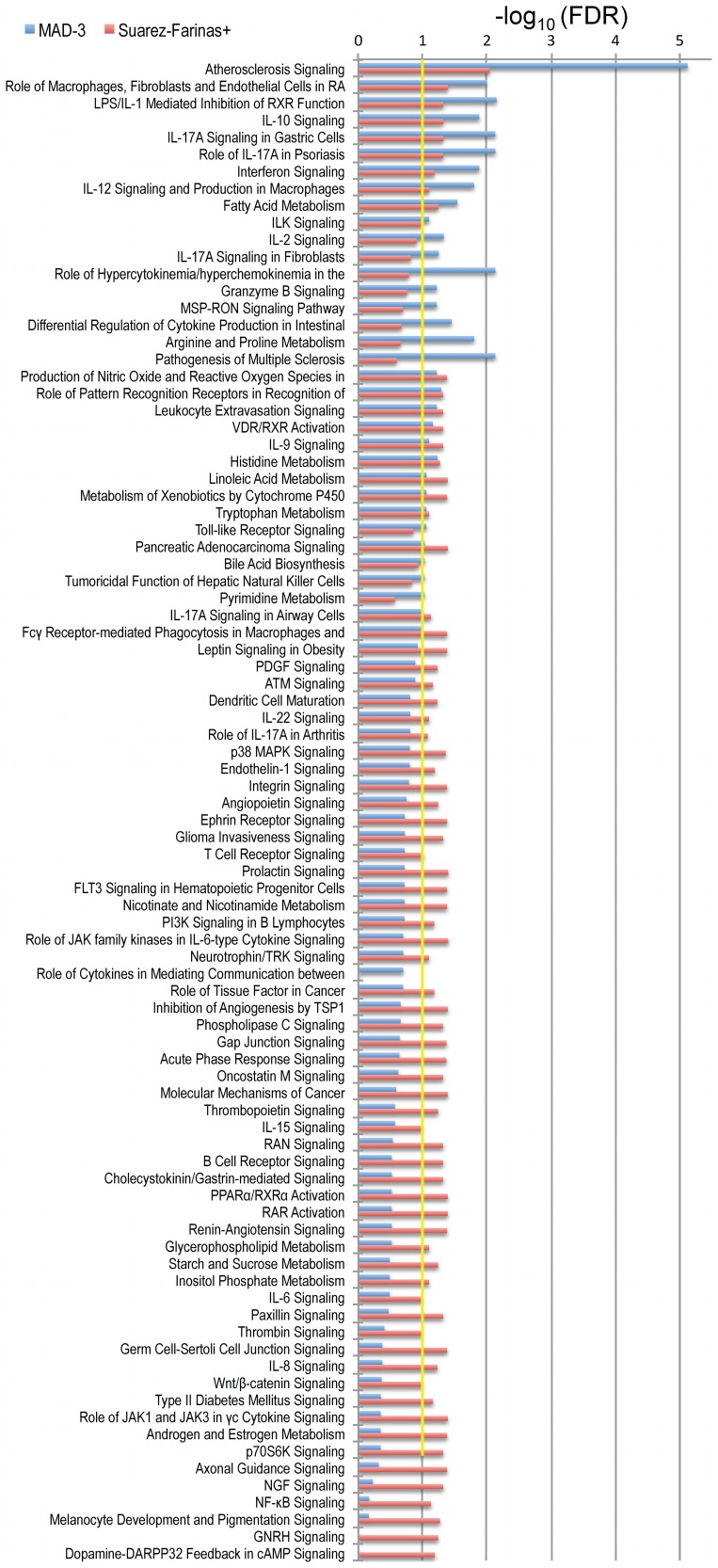
The cause of psoriasis, a common chronic inflammatory skin disease, is not fully understood. Microarray experiments have been widely used in recent years to identify genes associated with psoriasis pathology, by comparing expression levels of lesional (LS) with adjacent non-lesional (NL) skin. It is commonly observed that the differentially expressed genes (DEGs) differ greatly across experiments, due to variations introduced in the microarray experiment pipeline. Therefore, a statistically based meta-analytic approach, which combines the results of individual studies, is warranted. In this study, a meta-analysis was conducted on 5 microarray data sets, including 193 LS and NL pairs. We termed this the Meta-Analysis Derived (MAD) transcriptome. In "MAD-5" transcriptome, 677 genes were up-regulated and 443 were down-regulated in LS skin compared to NL skin. This represents a much larger set than the intersection of DEGs of these 5 studies, which consisted of 100 DEGs. We also analyzed 3 of the studies conducted on the Affymetrix hgu133plus2 chips and found a greater number of DEGs (1084 up- and 748 down-regulated). Top canonical pathways over-represented in the MAD transcriptome include Atherosclerosis Signaling and Fatty Acid Metabolism, while several "new" genes identified are involved in Cardiovascular Development and Lipid Metabolism. These findings highlight the relationship between psoriasis and systemic manifestations such as the metabolic syndrome and cardiovascular disease. Then, the Meta Threshold Gradient Descent Regularization (MTGDR) algorithm was used to select potential markers distinguishing LS and NL skin. The resulting set (20 genes) contained many genes that were part of the residual disease genomic profile (RDGP) or "molecular scar" after successful treatment, and also genes subject to differential methylation in LS tissues. To conclude, this MAD transcriptome yielded a reference list of reliable psoriasis DEGs, and represents a robust pool of candidates for further discovery of pathogenesis and treatment evaluation.
银屑病是一种常见的慢性炎症性皮肤病,其病因尚未完全阐明。近年来,通过比较病变(LS)和相邻非病变(NL)皮肤的表达水平,微阵列实验已广泛用于识别与银屑病病理学相关的基因。通常观察到,由于微阵列实验过程中的差异,差异表达基因(DEGs)在不同实验中差异很大。因此,需要进行基于统计学的荟萃分析方法,将个体研究的结果结合起来。在这项研究中,对包括 193 对 LS 和 NL 在内的 5 个微阵列数据集进行了荟萃分析。我们将其称为荟萃分析衍生(MAD)转录组。在“MAD-5”转录组中,LS 皮肤中 677 个基因上调,443 个基因下调。这比这 5 项研究的 DEGs 的交集要大得多,其中包括 100 个 DEGs。我们还分析了在 Affymetrix hgu133plus2 芯片上进行的 3 项研究,发现了更多的 DEGs(1084 个上调和 748 个下调)。MAD 转录组中过度表达的顶级经典途径包括动脉粥样硬化信号和脂肪酸代谢,而鉴定出的几个“新”基因参与心血管发育和脂质代谢。这些发现突出了银屑病与代谢综合征和心血管疾病等全身表现之间的关系。然后,使用 Meta Threshold Gradient Descent Regularization (MTGDR) 算法选择潜在的区分 LS 和 NL 皮肤的标记物。得到的集合(20 个基因)包含许多是成功治疗后残留疾病基因组谱(RDGP)或“分子疤痕”的一部分的基因,以及 LS 组织中差异甲基化的基因。总之,这个 MAD 转录组提供了一个可靠的银屑病 DEGs 参考列表,代表了进一步发现发病机制和治疗评估的强大候选者库。