Shiino Teiichiro
Infectious Diseases Surveillance Center, National Institute of Infectious Diseases Tokyo, Japan.
Front Microbiol. 2012 Jul 31;3:278. doi: 10.3389/fmicb.2012.00278. eCollection 2012.
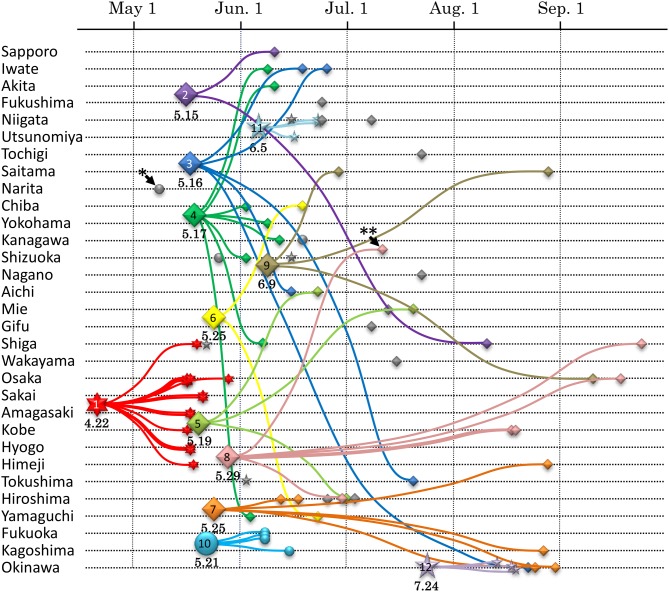
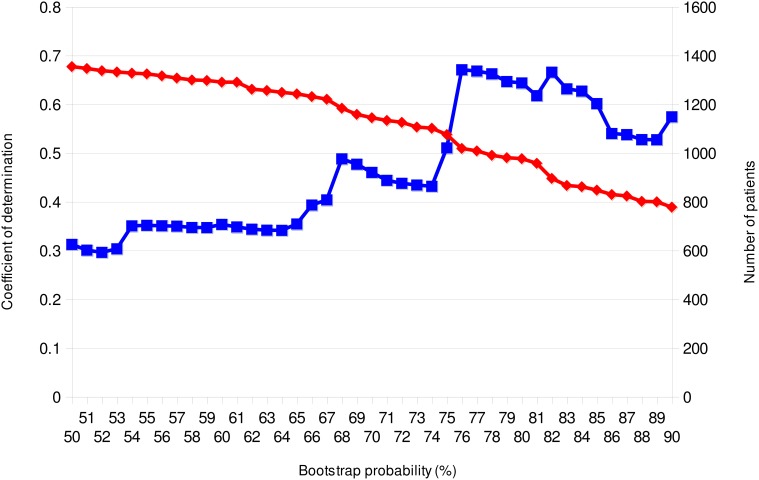
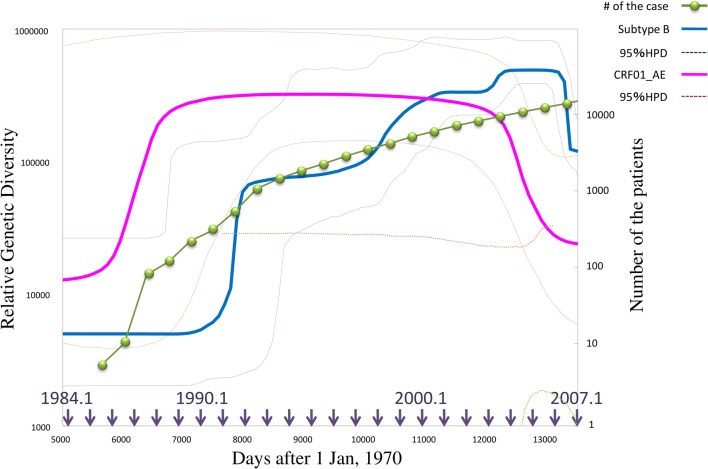
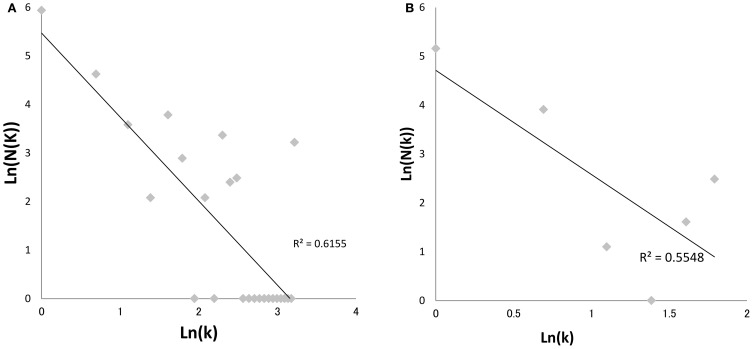
Viral infections by sexual and droplet transmission routes typically spread through a complex host-to-host contact network. Clarifying the transmission network and epidemiological parameters affecting the variations and dynamics of a specific pathogen is a major issue in the control of infectious diseases. However, conventional methods such as interview and/or classical phylogenetic analysis of viral gene sequences have inherent limitations and often fail to detect infectious clusters and transmission connections. Recent improvements in computational environments now permit the analysis of large datasets. In addition, novel analytical methods have been developed that serve to infer the evolutionary dynamics of virus genetic diversity using sample date information and sequence data. This type of framework, termed "phylodynamics," helps connect some of the missing links on viral transmission networks, which are often hard to detect by conventional methods of epidemiology. With sufficient number of sequences available, one can use this new inference method to estimate theoretical epidemiological parameters such as temporal distributions of the primary infection, fluctuation of the pathogen population size, basic reproductive number, and the mean time span of disease infectiousness. Transmission networks estimated by this framework often have the properties of a scale-free network, which are characteristic of infectious and social communication processes. Network analysis based on phylodynamics has alluded to various suggestions concerning the infection dynamics associated with a given community and/or risk behavior. In this review, I will summarize the current methods available for identifying the transmission network using phylogeny, and present an argument on the possibilities of applying the scale-free properties to these existing frameworks.
通过性传播和飞沫传播途径的病毒感染通常通过复杂的宿主间接触网络传播。阐明影响特定病原体变异和动态的传播网络及流行病学参数是传染病控制中的一个主要问题。然而,诸如访谈和/或对病毒基因序列进行经典系统发育分析等传统方法存在固有局限性,往往无法检测到感染集群和传播联系。计算环境的最新改进现在允许对大型数据集进行分析。此外,已经开发出新颖的分析方法,用于利用样本日期信息和序列数据推断病毒遗传多样性的进化动态。这种框架被称为“系统发育动力学”,有助于连接病毒传播网络上一些通常难以通过传统流行病学方法检测到的缺失环节。有了足够数量的可用序列,就可以使用这种新的推断方法来估计理论流行病学参数,如初次感染的时间分布、病原体种群大小的波动、基本繁殖数以及疾病传染性的平均时间跨度。通过这个框架估计的传播网络通常具有无标度网络的特性,这是感染和社会传播过程的特征。基于系统发育动力学的网络分析已经暗示了关于与特定社区和/或风险行为相关的感染动态的各种建议。在这篇综述中,我将总结目前利用系统发育识别传播网络的可用方法,并就将无标度特性应用于这些现有框架的可能性提出观点。