Volz Erik M, Romero-Severson Ethan, Leitner Thomas
Department of Infectious Disease Epidemiology, Imperial College London, London, UK.
Theoretical Biology and Biophysics, Group T-6, Los Alamos National Laboratory, Los Alamos.
Mol Biol Evol. 2017 May 1;34(5):1276-1288. doi: 10.1093/molbev/msx077.
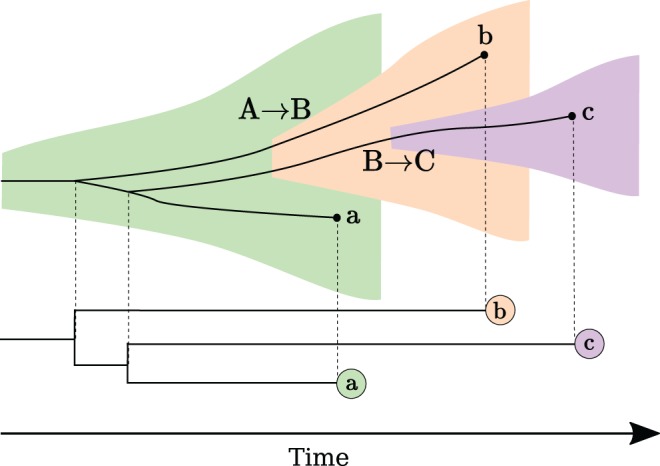
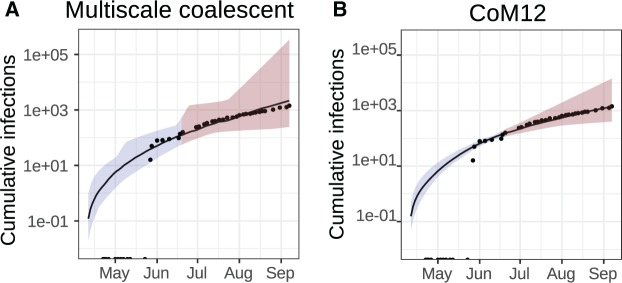
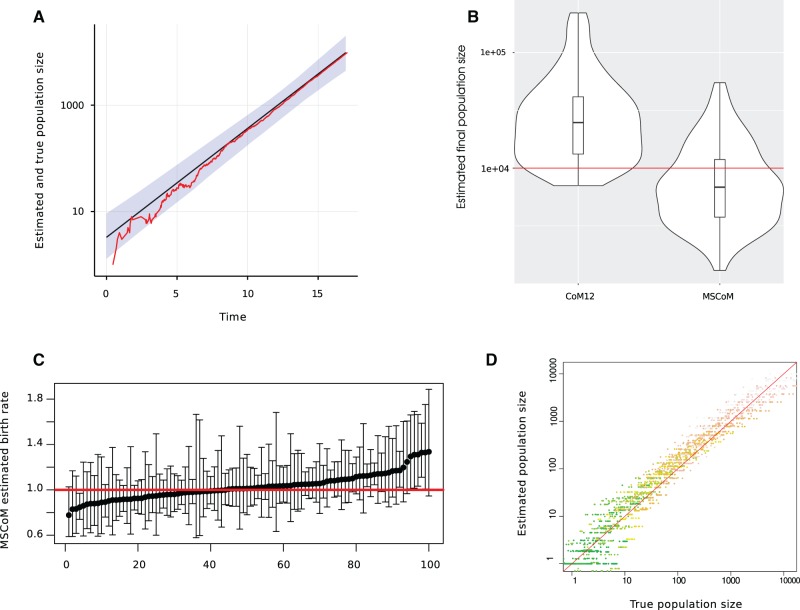
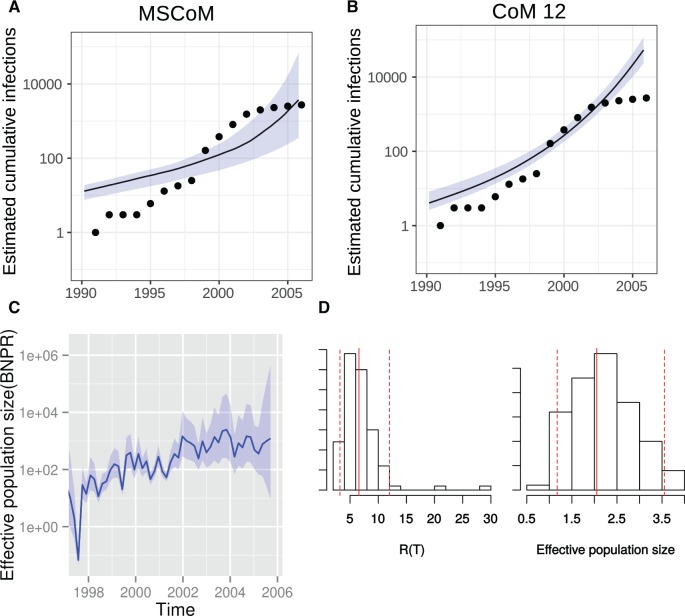
Within-host genetic diversity and large transmission bottlenecks confound phylodynamic inference of epidemiological dynamics. Conventional phylodynamic approaches assume that nodes in a time-scaled pathogen phylogeny correspond closely to the time of transmission between hosts that are ancestral to the sample. However, when hosts harbor diverse pathogen populations, node times can substantially pre-date infection times. Imperfect bottlenecks can cause lineages sampled in different individuals to coalesce in unexpected patterns. To address realistic violations of standard phylodynamic assumptions we developed a new inference approach based on a multi-scale coalescent model, accounting for nonlinear epidemiological dynamics, heterogeneous sampling through time, non-negligible genetic diversity of pathogens within hosts, and imperfect transmission bottlenecks. We apply this method to HIV-1 and Ebola virus (EBOV) outbreak sequence data, illustrating how and when conventional phylodynamic inference may give misleading results. Within-host diversity of HIV-1 causes substantial upwards bias in the number of infected hosts using conventional coalescent models, but estimates using the multi-scale model have greater consistency with reported number of diagnoses through time. In contrast, we find that within-host diversity of EBOV has little influence on estimated numbers of infected hosts or reproduction numbers, and estimates are highly consistent with the reported number of diagnoses through time. The multi-scale coalescent also enables estimation of within-host effective population size using single sequences from a random sample of patients. We find within-host population genetic diversity of HIV-1 p17 to be 2Nμ=0.012 (95% CI 0.0066-0.023), which is lower than estimates based on HIV envelope serial sequencing of individual patients.
宿主体内的基因多样性和较大的传播瓶颈混淆了流行病学动态的系统发育动力学推断。传统的系统发育动力学方法假设,在时间尺度上的病原体系统发育树中的节点与样本祖先宿主之间的传播时间密切对应。然而,当宿主携带多种病原体群体时,节点时间可能会大大早于感染时间。不完美的瓶颈可能导致在不同个体中采样的谱系以意想不到的模式合并。为了解决对标准系统发育动力学假设的实际违反问题,我们基于多尺度合并模型开发了一种新的推断方法,该模型考虑了非线性流行病学动态、随时间变化的异质采样、宿主体内病原体不可忽略的基因多样性以及不完美的传播瓶颈。我们将此方法应用于HIV-1和埃博拉病毒(EBOV)疫情序列数据,说明了传统系统发育动力学推断在何时以及如何可能给出误导性结果。使用传统合并模型时,HIV-1的宿主体内多样性会导致估计的感染宿主数量出现大幅向上偏差,但使用多尺度模型的估计在随时间变化时与报告的诊断数量具有更高的一致性。相比之下,我们发现EBOV的宿主体内多样性对估计的感染宿主数量或繁殖数影响很小,并且估计值与随时间变化的报告诊断数量高度一致。多尺度合并还能够使用来自患者随机样本的单序列估计宿主体内的有效种群大小。我们发现HIV-1 p17的宿主体内群体遗传多样性为2Nμ = 0.012(95%置信区间0.0066 - 0.023),这低于基于个体患者HIV包膜序列测序的估计值。