Pan Stephen, Caleshu Colleen A, Dunn Kyla E, Foti Marcia J, Moran Maura K, Soyinka Oretunlewa, Ashley Euan A
Stanford Center for Inherited Cardiovascular Disease, Stanford Hospital & Clinics, CA, USA.
Circ Cardiovasc Genet. 2012 Dec;5(6):602-10. doi: 10.1161/CIRCGENETICS.112.963421. Epub 2012 Oct 16.
The clinical significance of variants in genes associated with inherited cardiomyopathies can be difficult to determine because of uncertainty regarding population genetic variation and a surprising amount of tolerance of the genome even to loss-of-function variants. We hypothesized that genes associated with cardiomyopathy might be particularly resistant to the accumulation of genetic variation.
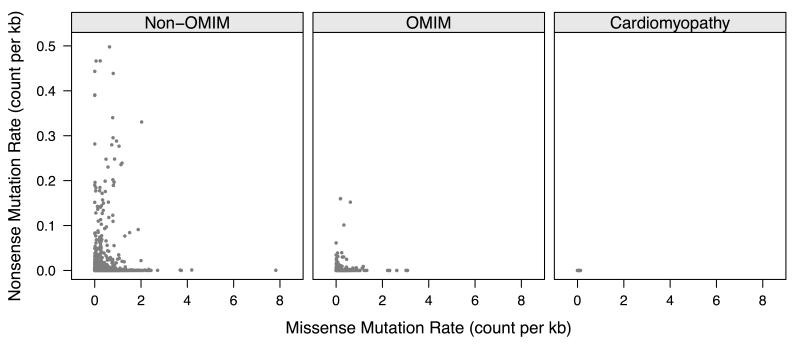
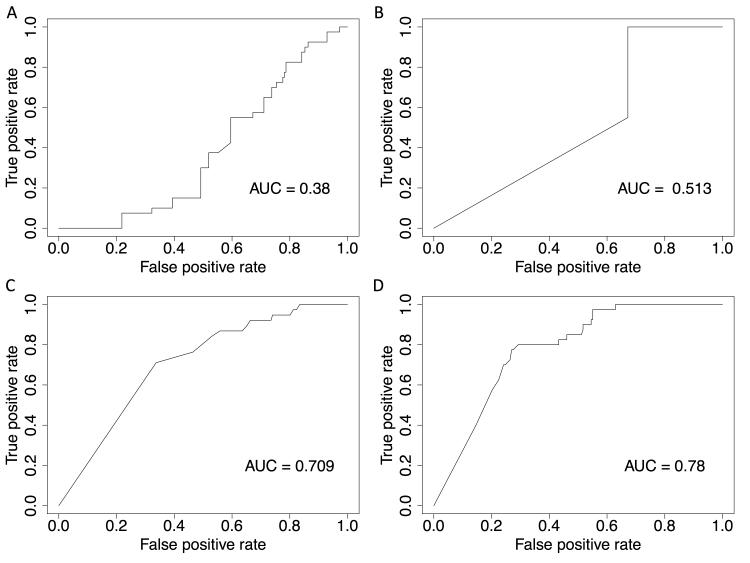
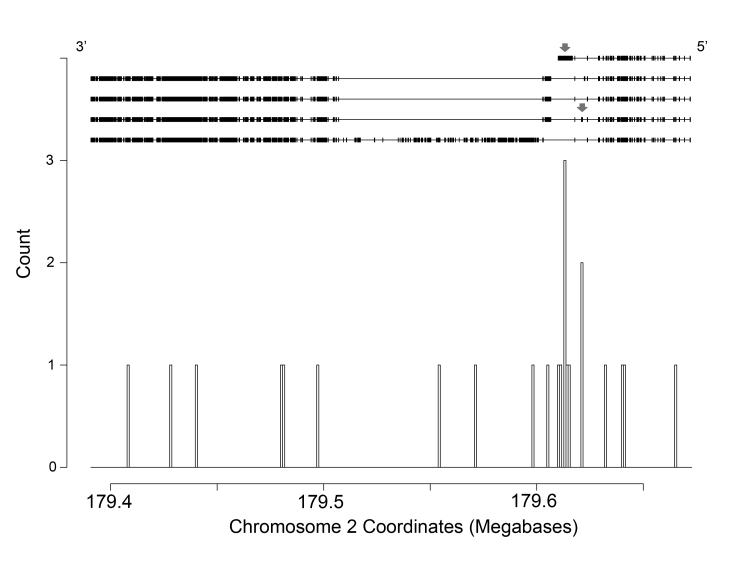
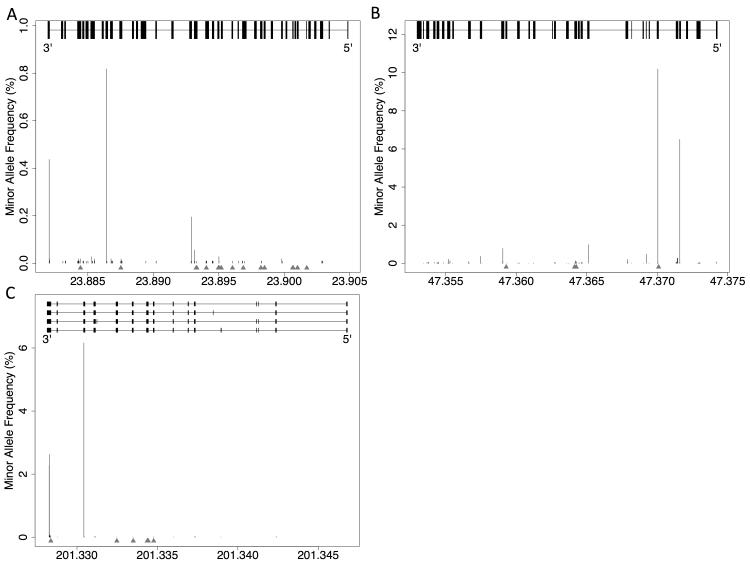
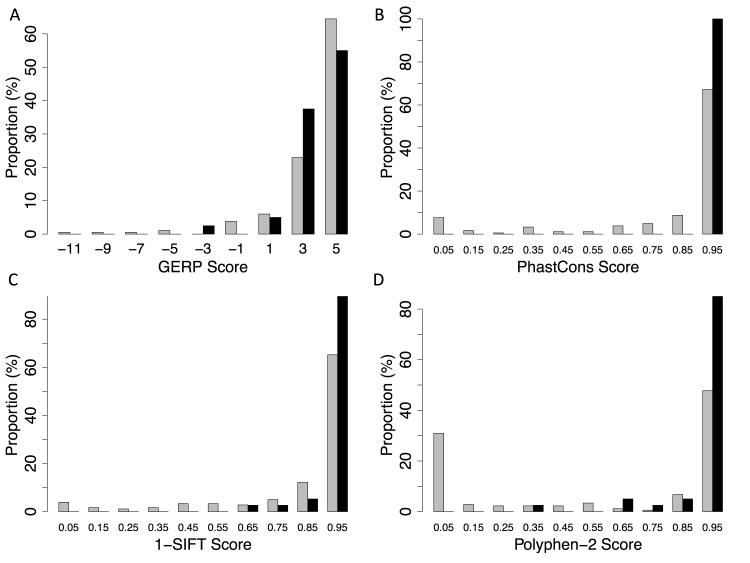
We analyzed the rates of single nucleotide genetic variation in all known genes from the exomes of >5000 individuals from the National Heart, Lung, and Blood Institute's Exome Sequencing Project, as well as the rates of structural variation from the Database of Genomic Variants. Most variants were rare, with over half unique to 1 individual. Cardiomyopathy-associated genes exhibited a rate of nonsense variants, about 96.1% lower than other Mendelian disease genes. We tested the ability of in silico algorithms to distinguish between a set of variants in MYBPC3, MYH7, and TNNT2 with strong evidence for pathogenicity and variants from the Exome Sequencing Project data. Algorithms based on conservation at the nucleotide level (genomic evolutionary rate profiling, PhastCons) did not perform as well as amino acid-level prediction algorithms (Polyphen-2, SIFT). Variants with strong evidence for disease causality were found in the Exome Sequencing Project data at prevalence higher than expected.
Genes associated with cardiomyopathy carry very low rates of population variation. The existence in population data of variants with strong evidence for pathogenicity suggests that even for Mendelian disease genetics, a probabilistic weighting of multiple variants may be preferred over the single gene causality model.
由于人群遗传变异的不确定性以及基因组对功能丧失变异的惊人耐受性,与遗传性心肌病相关基因变异的临床意义可能难以确定。我们推测与心肌病相关的基因可能对遗传变异的积累具有特别的抗性。
我们分析了来自美国国立心肺血液研究所外显子测序项目中5000多名个体外显子组中所有已知基因的单核苷酸遗传变异率,以及来自基因组变异数据库的结构变异率。大多数变异是罕见的,超过一半是某个个体所特有的。与心肌病相关的基因表现出无义变异率,比其他孟德尔疾病基因低约96.1%。我们测试了计算机算法区分MYBPC3、MYH7和TNNT2中一组具有强致病性证据的变异与外显子测序项目数据中的变异的能力。基于核苷酸水平保守性的算法(基因组进化速率分析,PhastCons)的表现不如氨基酸水平预测算法(Polyphen-2、SIFT)。在外显子测序项目数据中发现具有强疾病因果关系证据的变异,其流行率高于预期。
与心肌病相关的基因携带的人群变异率非常低。人群数据中存在具有强致病性证据的变异表明,即使对于孟德尔疾病遗传学,多个变异的概率加权可能比单基因因果模型更可取。