Department of Cancer Prevention and Controls, Roswell Park Cancer Institute, Buffalo, New York, United States of America.
PLoS One. 2012;7(11):e47962. doi: 10.1371/journal.pone.0047962. Epub 2012 Nov 2.
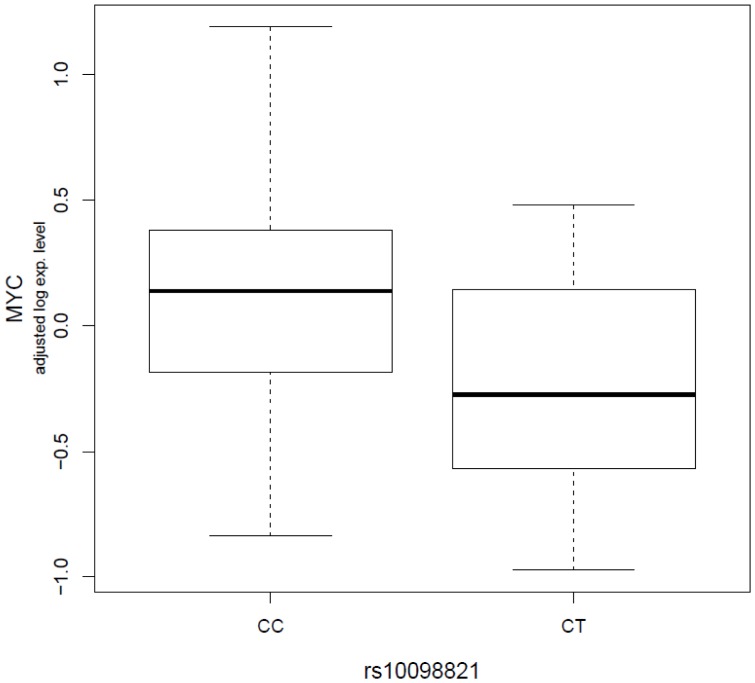
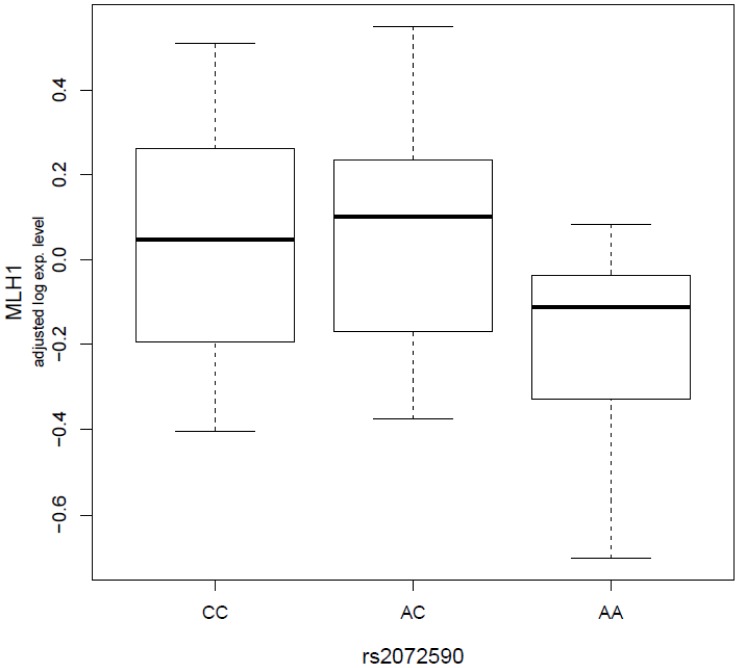
Functional genetic variations play important roles in shaping phenotypic differences among individuals through affecting gene expression, and thus, very likely to influence disease susceptibility, such as cancer susceptibility. One critical question in this era of post-genome wide association studies (GWAS) is how to assess the functional significance of the genetic variations identified from GWAS. In the current study, with lymphoblastoid cell lines (LCLs) from 74 non-related women with familial ovarian cancer and 47 unrelated controls matched on gender and race, we explored the associations between seven ovarian cancer risk variants identified from GWAS (rs3814113 on 9p22.2, rs2072590 on 2q31, rs2665390 on 3q25, rs10088218, rs1516982, rs10098821 on 8q24.21, and rs2363956 on 19p13) and whole genome mRNA expression profiles. We observed 95 significant trans-associations at a permutation level of 0.001. Compared to the other risk variants, rs10088218, rs1516982, and rs10098821 on 8q24.21 had the greatest number of significant associations (25, 16, and 38, respectively). Two possible cis-associations were observed between rs10098821 and c-Myc, and rs2072590 and HS.565379 (Permutated P = 0.0198 and 0.0399, respectively). Pathway enrichment analysis showed that several key biological pathways, such as cell cycle (P = 2.59×10(-06)), etc, were significantly overrepresented. Further characterization of significant associations between mRNAs and risk alleles might facilitate understanding the functions of GWAS discovered risk alleles in the genetic etiology of ovarian cancer.
功能性遗传变异通过影响基因表达在塑造个体表型差异方面发挥着重要作用,因此很可能会影响疾病易感性,如癌症易感性。在后全基因组关联研究(GWAS)时代,一个关键问题是如何评估从 GWAS 中鉴定出的遗传变异的功能意义。在这项研究中,我们使用来自 74 名非相关卵巢癌家族患者的淋巴母细胞系(LCL)和 47 名性别和种族匹配的无关对照,探索了从 GWAS 中鉴定出的七个卵巢癌风险变异(rs3814113 在 9p22.2 上,rs2072590 在 2q31 上,rs2665390 在 3q25 上,rs10088218、rs1516982、rs10098821 在 8q24.21 上,rs2363956 在 19p13 上)与全基因组 mRNA 表达谱之间的关联。我们在置换水平为 0.001 的情况下观察到了 95 个显著的跨关联。与其他风险变异相比,rs10088218、rs1516982 和 rs10098821 在 8q24.21 上有最多的显著关联(分别为 25、16 和 38 个)。在 rs10098821 和 c-Myc 之间以及 rs2072590 和 HS.565379 之间观察到两个可能的顺式关联(置换 P=0.0198 和 0.0399)。途径富集分析显示,几个关键的生物学途径,如细胞周期(P=2.59×10(-06))等,显著过表达。进一步描述 mRNAs 和风险等位基因之间的显著关联可能有助于理解 GWAS 发现的风险等位基因在卵巢癌遗传病因学中的功能。