Department of Biological Sciences, Michigan Technological University, Houghton, Michigan, United States of America.
PLoS One. 2012;7(11):e49331. doi: 10.1371/journal.pone.0049331. Epub 2012 Nov 8.
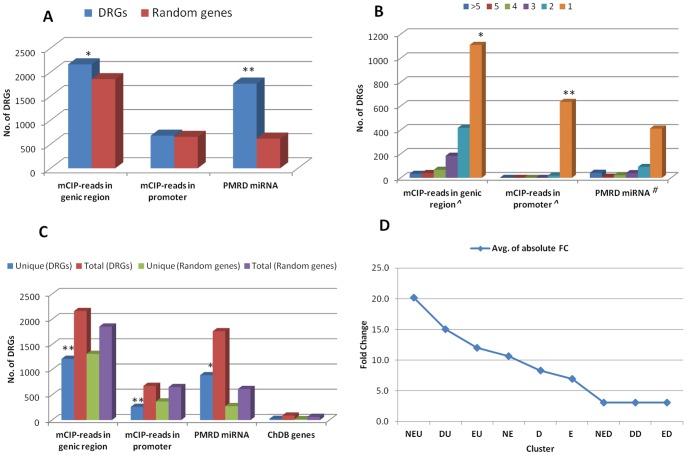
Drought stress response is a complex trait regulated at multiple levels. Changes in the epigenetic and miRNA regulatory landscape can dramatically alter the outcome of a stress response. However, little is known about the scope and extent of these regulatory factors on drought related cellular processes and functions. To this end, we selected a list of 5468 drought responsive genes (DRGs) of rice identified in multiple microarray studies and mapped the DNA methylation regions found in a genome wide methylcytosine immunoprecipitation and sequencing (mCIP-Seq) study to their genic and promoter regions, identified the chromatin remodeling genes and the genes that are targets of miRNAs. We found statistically significant enrichment of DNA methylation reads and miRNA target sequences in DRGs compared to a random set of genes. About 75% of the DRGs annotated to be involved in chromatin remodeling were downregulated. We found one-third of the DRGs are targeted by two-thirds of all known/predicted miRNAs in rice which include many transcription factors targeted by more than five miRNAs. Clustering analysis of the DRGs with epigenetic and miRNA features revealed, upregulated cluster was enriched in drought tolerance mechanisms while the downregulated cluster was enriched in drought resistance mechanisms evident by their unique gene ontologies (GOs), protein-protein interactions (PPIs), specific transcription factors, protein domains and metabolic pathways. Further, we analyzed the proteome of two weeks old young rice plants treated with a global demethylating agent, 5-azacytidine (5-azaC), subjected to drought stress and identified 56 protein spots that are differentially expressed. Out of the 56 spots, 35 were differently expressed in the sample with both demethylation and drought stress treatments and 28 (50%) were part of DRGs considered in the bioinformatic analysis.
干旱胁迫响应是一个受多个层次调节的复杂性状。表观遗传和 miRNA 调控景观的变化可以显著改变胁迫响应的结果。然而,对于这些调控因子对与干旱相关的细胞过程和功能的影响范围和程度知之甚少。为此,我们选择了在多个微阵列研究中鉴定的水稻 5468 个干旱响应基因 (DRGs) 的列表,并将在全基因组甲基胞嘧啶免疫沉淀和测序 (mCIP-Seq) 研究中发现的 DNA 甲基化区域映射到它们的基因和启动子区域,鉴定了染色质重塑基因和 miRNA 的靶基因。我们发现与随机基因集相比,DRGs 中的 DNA 甲基化读取和 miRNA 靶序列存在统计学上的显著富集。大约 75%注释为参与染色质重塑的 DRGs 下调。我们发现三分之一的 DRGs 是水稻中三分之二已知/预测 miRNA 的靶基因,其中包括许多被超过五个 miRNA 靶向的转录因子。对具有表观遗传和 miRNA 特征的 DRGs 进行聚类分析表明,上调簇富集了干旱耐受机制,而下调簇富集了干旱抵抗机制,这可以从它们独特的基因本体 (GO)、蛋白质-蛋白质相互作用 (PPI)、特定转录因子、蛋白质结构域和代谢途径中看出。此外,我们分析了用全局去甲基化剂 5-氮杂胞苷 (5-azaC) 处理两周大的幼稻植株的蛋白质组,然后进行干旱胁迫处理,鉴定出 56 个差异表达的蛋白质斑点。在 56 个斑点中,有 35 个在去甲基化和干旱胁迫处理的样本中表达不同,其中 28 (50%) 是生物信息学分析中考虑的 DRGs 的一部分。