Thomson Reuters, IP & Science, 5901 Priestly Dr., #200, Carlsbad, CA 92008, USA.
BMC Bioinformatics. 2012;13 Suppl 16(Suppl 16):S13. doi: 10.1186/1471-2105-13-S16-S13. Epub 2012 Nov 5.
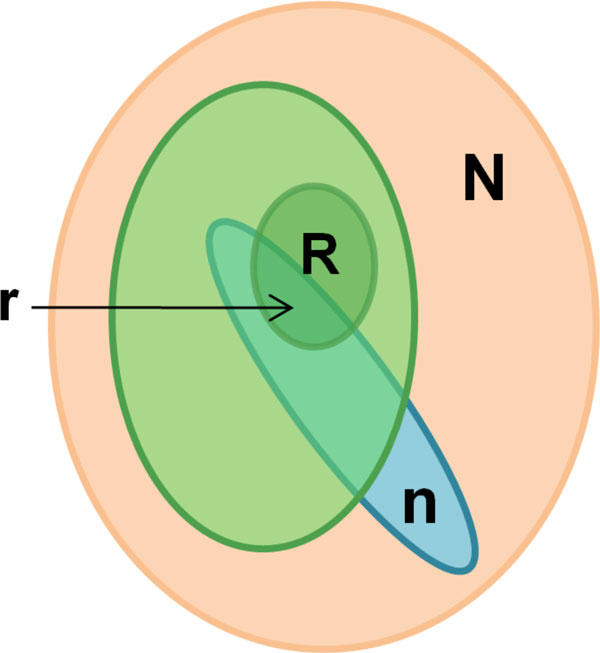
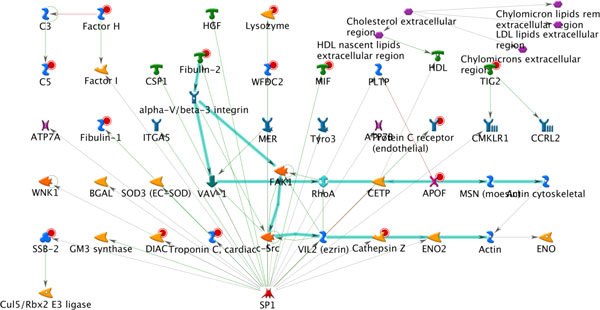
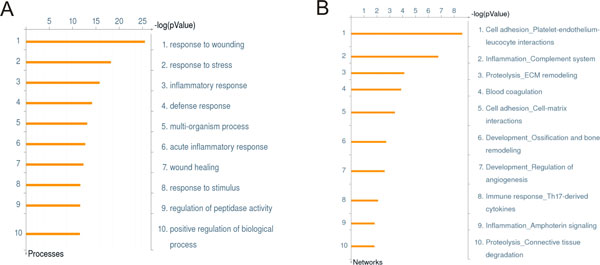
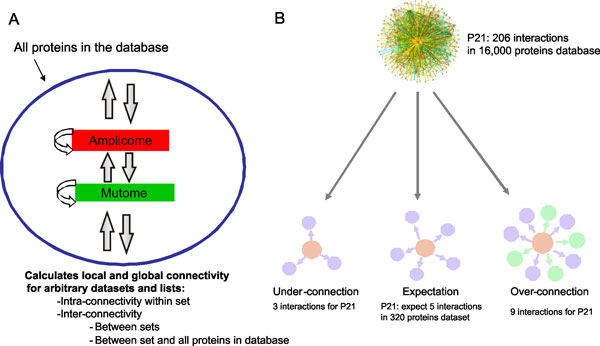
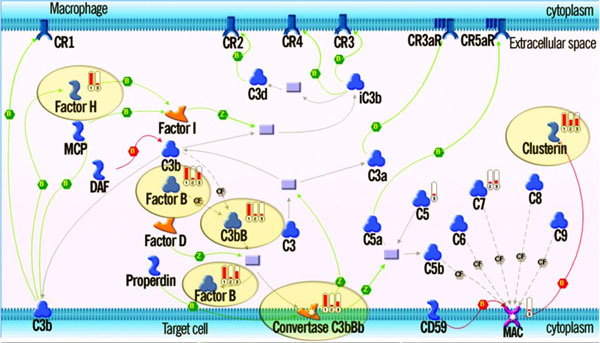
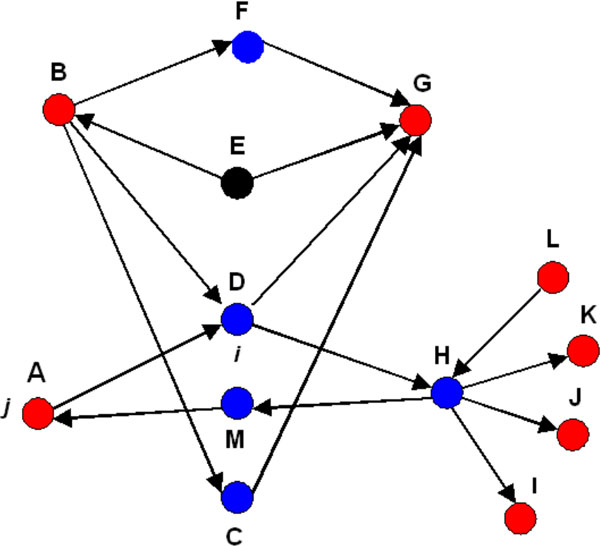
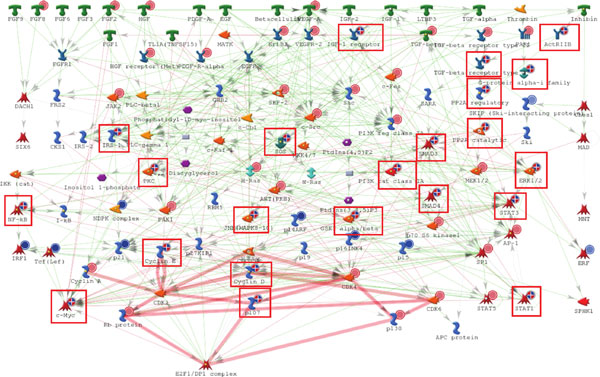
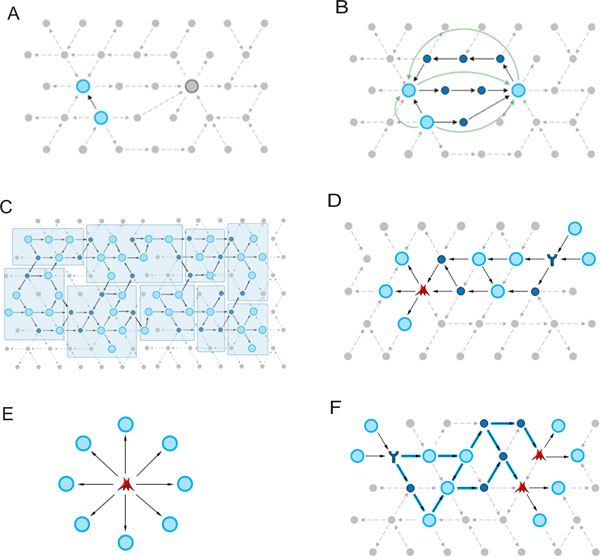
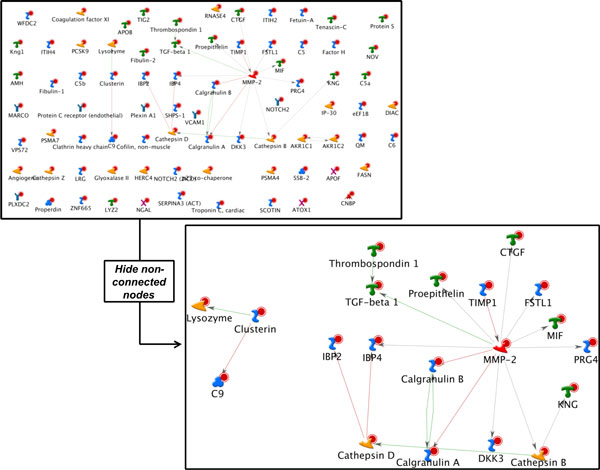
As it is the case with any OMICs technology, the value of proteomics data is defined by the degree of its functional interpretation in the context of phenotype. Functional analysis of proteomics profiles is inherently complex, as each of hundreds of detected proteins can belong to dozens of pathways, be connected in different context-specific groups by protein interactions and regulated by a variety of one-step and remote regulators. Knowledge-based approach deals with this complexity by creating a structured database of protein interactions, pathways and protein-disease associations from experimental literature and a set of statistical tools to compare the proteomics profiles with this rich source of accumulated knowledge. Here we describe the main methods of ontology enrichment, interactome topology and network analysis applied on a comprehensive, manually curated and semantically consistent knowledge source MetaBase and demonstrate several case studies in different disease areas.
与任何 OMICs 技术一样,蛋白质组学数据的价值取决于其在表型背景下功能解释的程度。蛋白质组学图谱的功能分析本质上很复杂,因为数百种检测到的蛋白质中的每一种都可能属于数十种途径,通过蛋白质相互作用连接到不同特定于上下文的组中,并受到各种一步和远程调节剂的调节。基于知识的方法通过从实验文献中创建蛋白质相互作用、途径和蛋白质-疾病关联的结构化数据库以及一组统计工具来处理这种复杂性,用于将蛋白质组学图谱与积累的丰富知识库进行比较。在这里,我们描述了本体富集、互作组拓扑和网络分析的主要方法,应用于全面的、手动整理和语义一致的知识库 MetaBase,并在不同疾病领域展示了几个案例研究。