Department of Biochemistry and Molecular Biophysics, Washington University in St. Louis, St. Louis, Missouri 63110, USA.
J Comput Chem. 2013 Apr 5;34(9):739-49. doi: 10.1002/jcc.23190. Epub 2012 Dec 5.
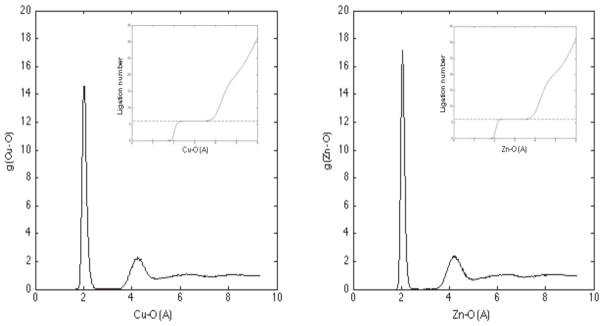
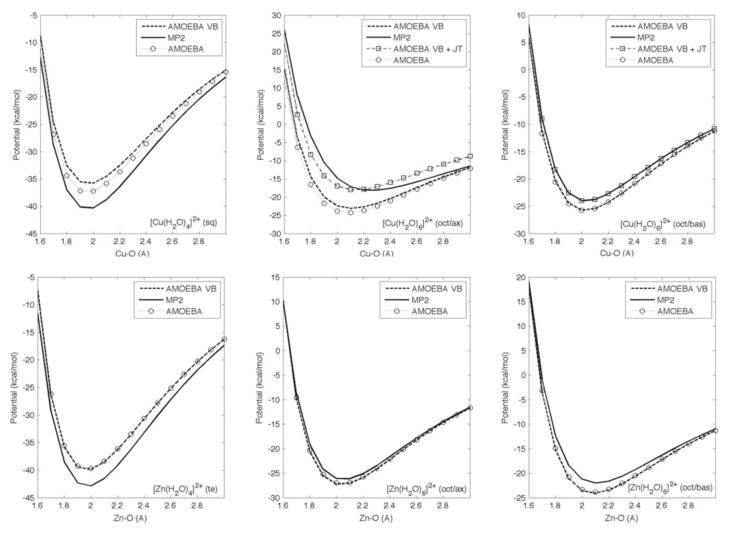
A general molecular mechanics (MM) model for treating aqueous Cu(2+) and Zn(2+) ions was developed based on valence bond (VB) theory and incorporated into the atomic multipole optimized energetics for biomolecular applications (AMOEBA) polarizable force field. Parameters were obtained by fitting MM energies to that computed by ab initio methods for gas-phase tetra- and hexa-aqua metal complexes. Molecular dynamics (MD) simulations using the proposed AMOEBA-VB model were performed for each transition metal ion in aqueous solution, and solvent coordination was evaluated. Results show that the AMOEBA-VB model generates the correct square-planar geometry for gas-phase tetra-aqua Cu(2+) complex and improves the accuracy of MM model energetics for a number of ligation geometries when compared to quantum mechanical (QM) computations. On the other hand, both AMOEBA and AMOEBA-VB generate results for Zn(2+)-water complexes in good agreement with QM calculations. Analyses of the MD trajectories revealed a six-coordination first solvation shell for both Cu(2+) and Zn(2+) ions in aqueous solution, with ligation geometries falling in the range reported by previous studies.
基于价键(VB)理论,开发了一种用于处理水溶液中 Cu(2+) 和 Zn(2+) 离子的通用分子力学(MM)模型,并将其纳入用于生物分子应用的原子多极优化能量学(AMOEBA)极化力场。通过将 MM 能量拟合到气相四水和六水金属配合物的从头算方法计算的能量,获得了参数。使用所提出的 AMOEBA-VB 模型对水溶液中的每个过渡金属离子进行了分子动力学(MD)模拟,并评估了溶剂配位。结果表明,AMOEBA-VB 模型为气相四水合 Cu(2+) 配合物生成了正确的平面正方形几何形状,并与量子力学(QM)计算相比,提高了许多配体几何形状的 MM 模型能量学的准确性。另一方面,AMOEBA 和 AMOEBA-VB 都为 Zn(2+)-水配合物生成了与 QM 计算结果相符的结果。对 MD 轨迹的分析表明,Cu(2+) 和 Zn(2+) 离子在水溶液中的第一溶剂化壳均为六配位,配体几何形状落在先前研究报告的范围内。