Laboratory of Experimental Medicine, Universite Libre de Bruxelles, Brussels, Belgium.
Ann Neurol. 2012 Dec;72(6):971-82. doi: 10.1002/ana.23698.
Friedreich ataxia (FRDA) is an autosomal recessive neurodegenerative disease caused in almost all cases by homozygosity for a GAA trinucleotide repeat expansion in the frataxin gene. Frataxin is a mitochondrial protein involved in iron homeostasis. FRDA patients have a high prevalence of diabetes, the pathogenesis of which is not known. We aimed to evaluate the relative contribution of insulin resistance and β-cell failure and the pathogenic mechanisms involved in FRDA diabetes.
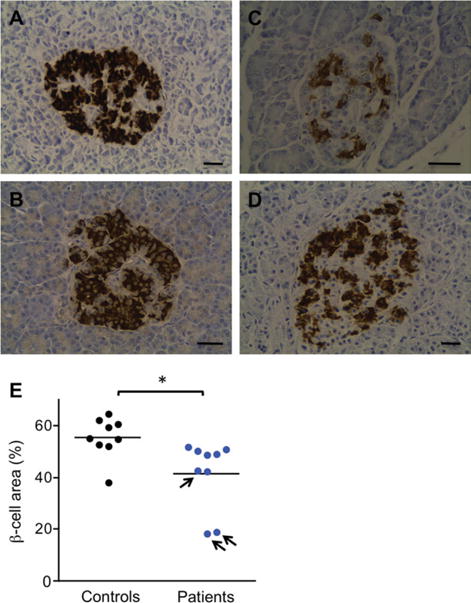
Forty-one FRDA patients, 26 heterozygous carriers of a GAA expansion, and 53 controls underwent oral and intravenous glucose tolerance tests. β-Cell proportion was quantified in postmortem pancreas sections from 9 unrelated FRDA patients. Using an in vitro disease model, we studied how frataxin deficiency affects β-cell function and survival.
FRDA patients had increased abdominal fat and were insulin resistant. This was not compensated for by increased insulin secretion, resulting in a markedly reduced disposition index, indicative of pancreatic β-cell failure. Loss of glucose tolerance was driven by β-cell dysfunction, which correlated with abdominal fatness. In postmortem pancreas sections, pancreatic islets of FRDA patients had a lower β-cell content. RNA interference-mediated frataxin knockdown impaired glucose-stimulated insulin secretion and induced apoptosis in rat β cells and human islets. Frataxin deficiency sensitized β cells to oleate-induced and endoplasmic reticulum stress-induced apoptosis, which could be prevented by the incretins glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide.
Pancreatic β-cell dysfunction is central to diabetes development in FRDA as a result of mitochondrial dysfunction and higher sensitivity to metabolic and endoplasmic reticulum stress-induced β-cell death.
弗里德赖希共济失调(FRDA)是一种常染色体隐性神经退行性疾病,几乎所有病例都是由于铁蛋白基因中 GAA 三核苷酸重复扩增导致纯合子所致。铁蛋白是一种参与铁稳态的线粒体蛋白。FRDA 患者糖尿病患病率很高,但发病机制尚不清楚。我们旨在评估胰岛素抵抗和β细胞衰竭的相对贡献以及 FRDA 糖尿病涉及的发病机制。
41 名 FRDA 患者、26 名 GAA 扩增杂合子携带者和 53 名对照者接受了口服和静脉葡萄糖耐量试验。从 9 名无关 FRDA 患者的尸检胰腺切片中定量β细胞比例。使用体外疾病模型,我们研究了铁蛋白缺乏如何影响β细胞功能和存活。
FRDA 患者腹部脂肪增加且存在胰岛素抵抗。这并没有通过增加胰岛素分泌来补偿,导致处置指数明显降低,表明β细胞衰竭。葡萄糖耐量的丧失是由β细胞功能障碍驱动的,这与腹部肥胖有关。在尸检胰腺切片中,FRDA 患者的胰岛β细胞含量较低。RNA 干扰介导的铁蛋白敲低会损害葡萄糖刺激的胰岛素分泌并诱导大鼠β细胞和人胰岛细胞凋亡。铁蛋白缺乏使β细胞对油酸盐诱导和内质网应激诱导的细胞凋亡更为敏感,而肠降血糖素 GLP-1 和葡萄糖依赖性胰岛素释放肽可以预防这种凋亡。
由于线粒体功能障碍和对代谢和内质网应激诱导的β细胞死亡的敏感性增加,β细胞功能障碍是 FRDA 糖尿病发展的核心。