Scarpelli Mauro, Russignan Anna, Zombor Melinda, Bereczki Csaba, Zappini Francesca, Buono Romina, Bax Bridget E, Padovani Alessandro, Tonin Paola, Filosto Massimiliano
Clinical Neurology, Department of Neurological, Neuropsychological, Morphological and Movement Sciences, University of Verona, Verona, Italy.
Case Rep Neurol. 2012 Sep;4(3):248-53. doi: 10.1159/000346260. Epub 2012 Dec 20.
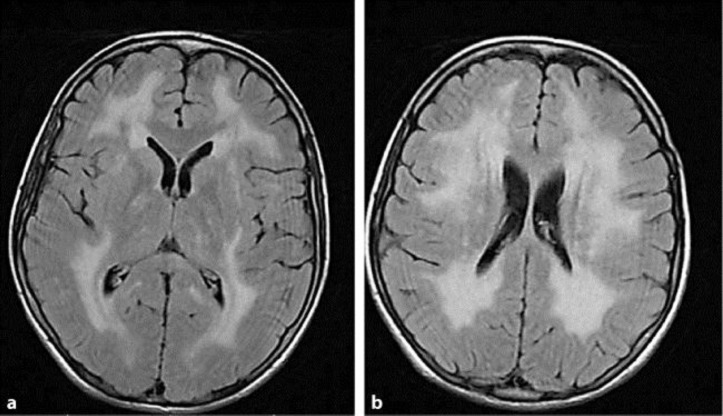
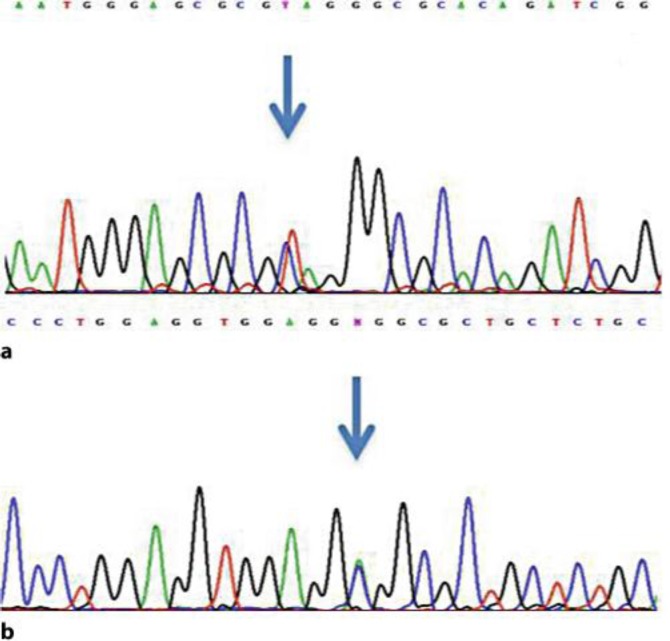
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is a devastating autosomal recessive disorder due to mutations in TYMP, which cause loss of function of thymidine phosphorylase (TP), nucleoside accumulation in plasma and tissues and mitochondrial dysfunction. The clinical picture includes progressive gastrointestinal dysmotility, cachexia, ptosis and ophthalmoparesis, peripheral neuropathy and diffuse leukoencephalopathy, which usually lead to death in early adulthood. Therapeutic options are currently available in clinical practice (allogeneic hematopoietic stem cell transplantation and carrier erythrocyte entrapped TP therapy) and newer, promising therapies are expected in the near future. However, successful treatment is strictly related to early diagnosis. We report on an incomplete MNGIE phenotype in a young man harboring the novel heterozygote c.199 C>T (Q67X) mutation in exon 2, and the previously reported c.866 A>C (E289A) mutation in exon 7 in TYMP. The correct diagnosis was achieved many years after the onset of symptoms and unfortunately, the patient died soon after diagnosis because of multiorgan failure due to severe malnutrition and cachexia before any therapeutic option could be tried. To date, early diagnosis is essential to ensure that patients have the opportunity to be treated. MNGIE should be suspected in all patients who present with both gastrointestinal and nervous system involvement, even if the classical complete phenotype is lacking.
线粒体神经胃肠性脑肌病(MNGIE)是一种由TYMP基因突变引起的严重常染色体隐性疾病,该突变导致胸苷磷酸化酶(TP)功能丧失、血浆和组织中核苷蓄积以及线粒体功能障碍。临床症状包括进行性胃肠动力障碍、恶病质、上睑下垂和眼肌麻痹、周围神经病变以及弥漫性白质脑病,通常导致患者在成年早期死亡。目前临床实践中有治疗选择(同种异体造血干细胞移植和携带TP的红细胞疗法),预计在不久的将来会有更新的、有前景的疗法。然而,成功治疗与早期诊断密切相关。我们报告了一名年轻男性的不完全MNGIE表型,其在TYMP基因外显子2中携带新的杂合子c.199 C>T(Q67X)突变,以及之前报道的外显子7中的c.866 A>C(E289A)突变。在症状出现多年后才做出正确诊断,不幸的是,患者在诊断后不久因严重营养不良和恶病质导致的多器官衰竭死亡,此前未能尝试任何治疗方案。迄今为止,早期诊断对于确保患者有机会接受治疗至关重要。对于所有出现胃肠道和神经系统受累症状的患者,即使缺乏典型的完整表型,也应怀疑患有MNGIE。