Tawk Antonios, Hussein Kamarreddine Mohammed, Dagher Mona, Abboud Ghadi, Chams Mohamad, Ghandour-Hajj Fatmeh, Khoury Mounir, Farhat Said
Faculty of Medicine and Medical Sciences, University of Balamand, Beirut, Lebanon.
Department of Diagnostic Radiology, Saint George Hospital University Medical Center, University of Balamand, Beirut, Lebanon.
Case Rep Gastroenterol. 2020 Apr 2;14(1):124-130. doi: 10.1159/000506187. eCollection 2020 Jan-Apr.
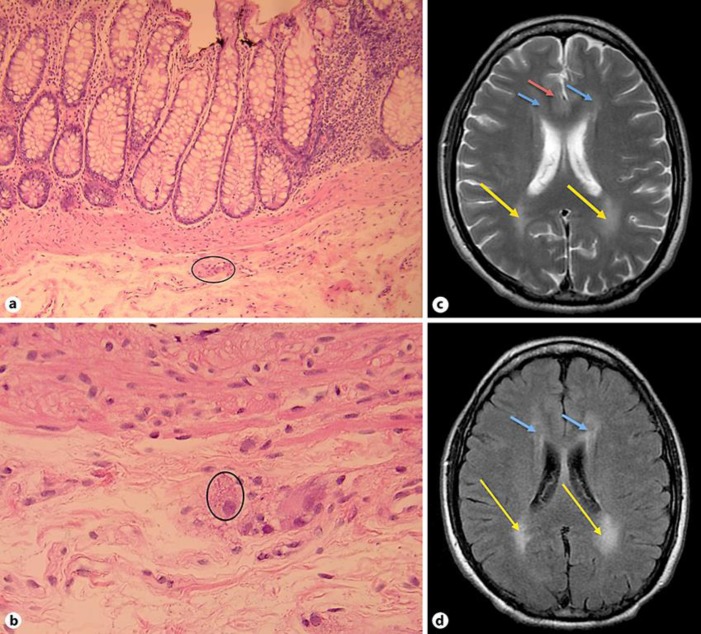
Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) is an autosomal recessive and fatal multisystem metabolic disorder. It presents with wide-ranging gastrointestinal and neurologic symptoms. It is caused by a mutation in the TYMP gene which impairs thymidine phosphorylase (TP) activity, therefore leading to the accumulation of thymidine and deoxyuridine in plasma and tissues. Thus, MNGIE can be diagnosed by findings of high levels of thymidine and deoxyuridine. Herein, we present the case of a 40-year-old male who presented with diarrhea, vomiting, and abdominal pain, severe weight loss, neurologic deficits, and distal motor weakness progressing over a period of 13 years. The combination of this broad clinical picture along with results of magnetic resonance imaging, electromyography, colonic biopsies, genetic testing, and elevated plasma and tissue thymidine and deoxyuridine levels confirmed the diagnosis of MNGIE. TYMP gene mutation impairs TP function. TP mutations in the nuclear DNA lead to mitochondrial DNA deletions causing mitochondrial failure and ultimately cell death. Treatment modalities are targeting the restoration of TP activity or aiming to decrease the high levels of thymidine and pyrimide. However, diagnosing this disease is still a challenge and often overdue. This patient's 13-year delay in diagnosis shows the importance of a complete neurological exam and muscle strength testing in patients with gastrointestinal symptoms. The diagnosis of MNGIE requires interdepartmental collaborative work for diagnosis delay prevention and for optimal patient care.
线粒体神经胃肠性脑肌病(MNGIE)是一种常染色体隐性遗传的致命性多系统代谢紊乱疾病。它表现出广泛的胃肠道和神经系统症状。它由TYMP基因突变引起,该突变会损害胸苷磷酸化酶(TP)的活性,从而导致血浆和组织中胸苷和脱氧尿苷的积累。因此,MNGIE可通过检测到高水平的胸苷和脱氧尿苷来诊断。在此,我们报告一例40岁男性患者,他在13年的时间里出现腹泻、呕吐、腹痛、严重体重减轻、神经功能缺损和远端运动无力。这种广泛的临床表现,结合磁共振成像、肌电图、结肠活检、基因检测以及血浆和组织中胸苷和脱氧尿苷水平升高的结果,确诊为MNGIE。TYMP基因突变会损害TP功能。核DNA中的TP突变会导致线粒体DNA缺失,从而引起线粒体功能衰竭并最终导致细胞死亡。治疗方法旨在恢复TP活性或降低高水平的胸苷和嘧啶。然而,诊断这种疾病仍然是一项挑战,而且往往会延误。该患者13年的诊断延迟表明,对于有胃肠道症状的患者,进行全面的神经系统检查和肌肉力量测试非常重要。MNGIE的诊断需要跨部门的协作,以预防诊断延迟并为患者提供最佳护理。