Biostatistics Program, School of Public Health, Louisiana State University Health Sciences Center, New Orleans, Louisiana, USA.
PLoS One. 2013;8(1):e54215. doi: 10.1371/journal.pone.0054215. Epub 2013 Jan 18.
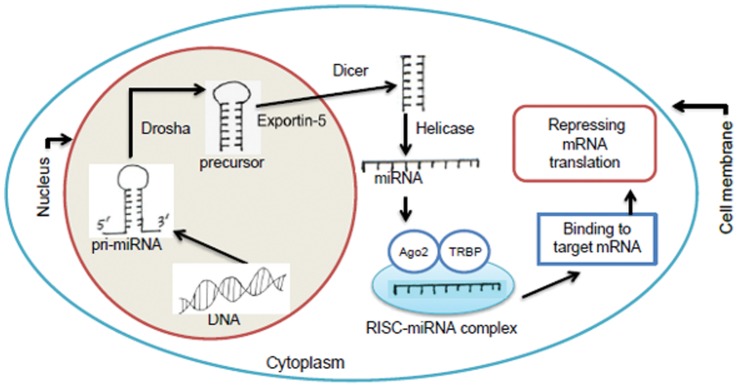
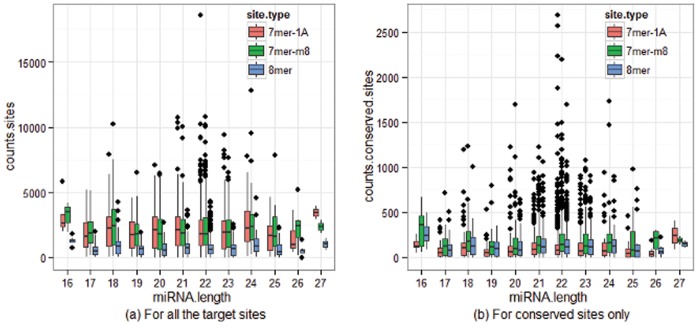
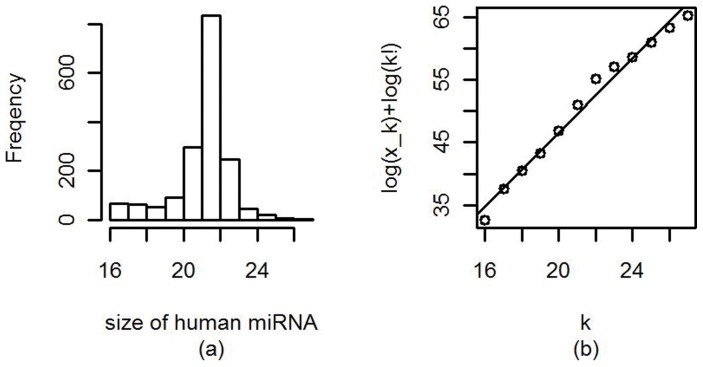
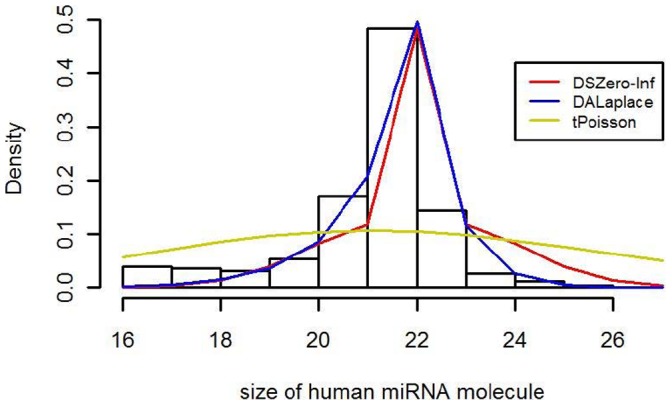
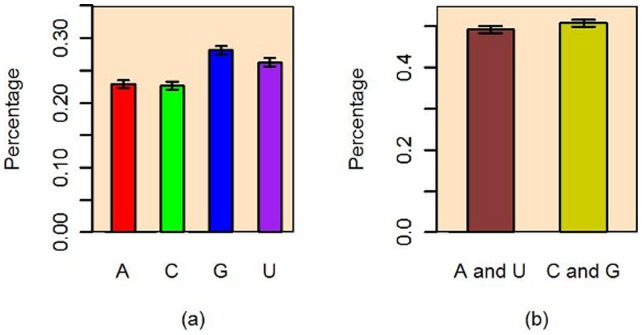
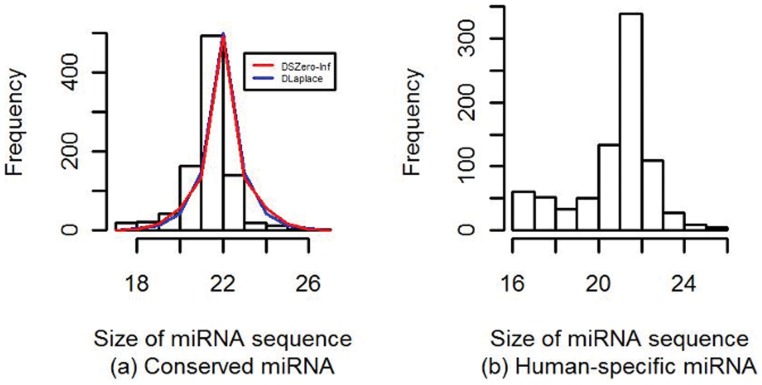
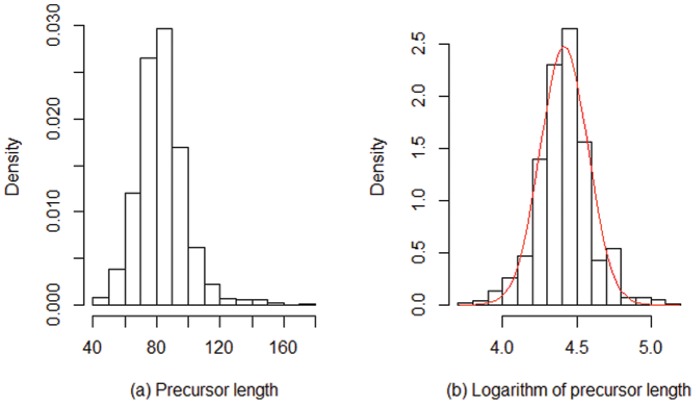
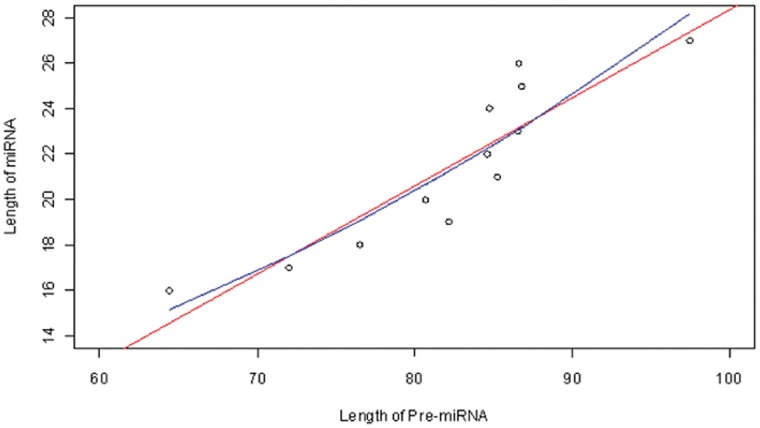
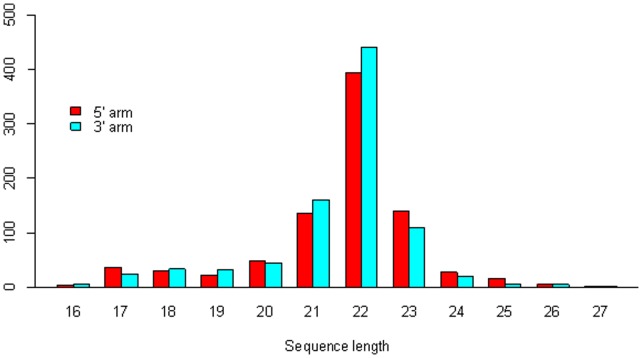
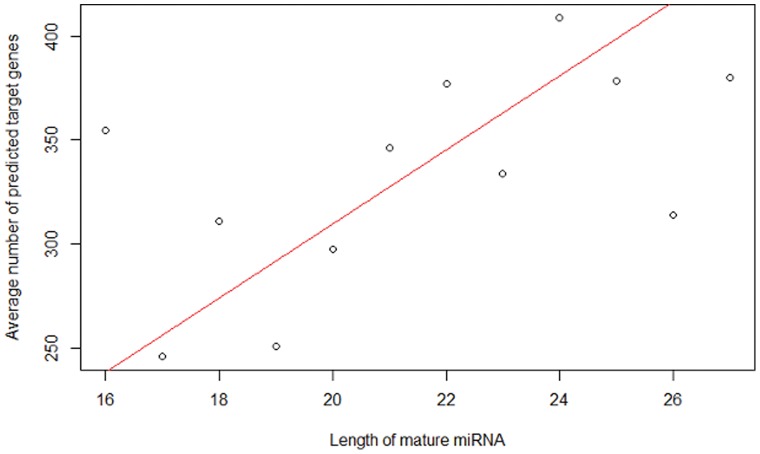
The count of the nucleotides in a cloned, short genomic sequence has become an important criterion to annotate such a sequence as a miRNA molecule. While the majority of human mature miRNA sequences consist of 22 nucleotides, there exists discrepancy in the characteristic lengths of the miRNA sequences. There is also a lack of systematic studies on such length distribution and on the biological factors that are related to or may affect this length. In this paper, we intend to fill this gap by investigating the sequence structure of human miRNA molecules using statistics tools. We demonstrate that the traditional discrete probability distributions do not model the length distribution of the human mature miRNAs well, and we obtain the statistical distribution model with a decent fit. We observe that the four nucleotide bases in a miRNA sequence are not randomly distributed, implying that possible structural patterns such as dinucleotide (trinucleotide or higher order) may exist. Furthermore, we study the relationships of this length distribution to multiple important factors such as evolutionary conservation, tumorigenesis, the length of precursor loop structures, and the number of predicted targets. The association between the miRNA sequence length and the distributions of target site counts in corresponding predicted genes is also presented. This study results in several novel findings worthy of further investigation that include: (1) rapid evolution introduces variation to the miRNA sequence length distribution; (2) miRNAs with extreme sequence lengths are unlikely to be cancer-related; and (3) the miRNA sequence length is positively correlated to the precursor length and the number of predicted target genes.
克隆的短基因组序列中的核苷酸数量计数已成为注释此类序列为 miRNA 分子的重要标准。虽然大多数人类成熟 miRNA 序列由 22 个核苷酸组成,但 miRNA 序列的特征长度存在差异。此外,对于这种长度分布以及与长度相关或可能影响长度的生物学因素,也缺乏系统的研究。在本文中,我们拟通过使用统计工具研究人类 miRNA 分子的序列结构来填补这一空白。我们证明,传统的离散概率分布并不能很好地模拟人类成熟 miRNA 的长度分布,我们获得了具有良好拟合度的统计分布模型。我们观察到 miRNA 序列中的四个核苷酸碱基不是随机分布的,这表明可能存在结构模式,如二核苷酸(三核苷酸或更高阶)。此外,我们研究了这种长度分布与多个重要因素(如进化保守性、肿瘤发生、前体环结构的长度和预测靶标的数量)之间的关系。还研究了 miRNA 序列长度与相应预测基因中靶标计数分布之间的关系。这项研究产生了一些值得进一步研究的新发现,包括:(1)快速进化导致 miRNA 序列长度分布的变化;(2)序列长度极端的 miRNA 不太可能与癌症有关;(3)miRNA 序列长度与前体长度和预测靶基因的数量呈正相关。