La Jolla Institute for Allergy and Immunology, La Jolla, California, United States of America.
PLoS One. 2013;8(1):e54359. doi: 10.1371/journal.pone.0054359. Epub 2013 Jan 30.
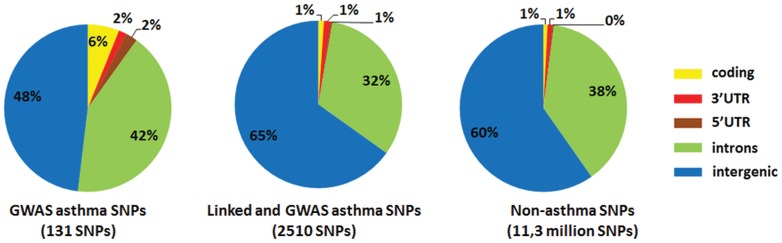
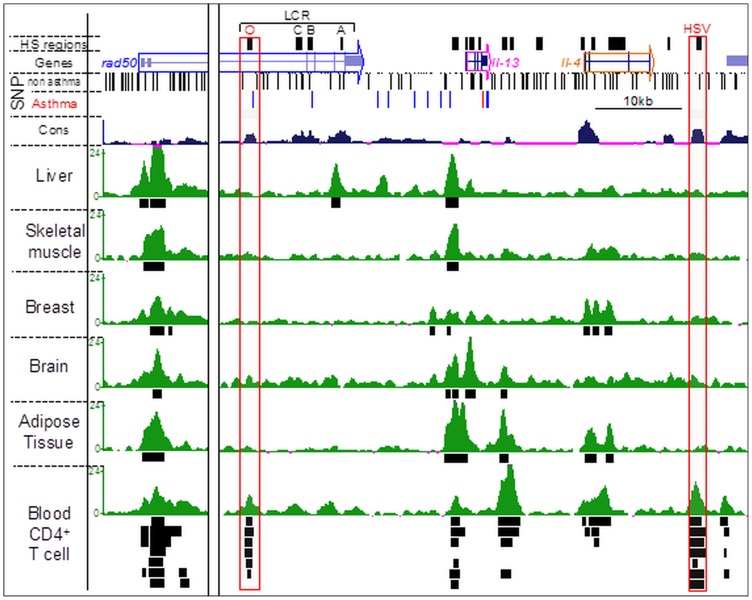
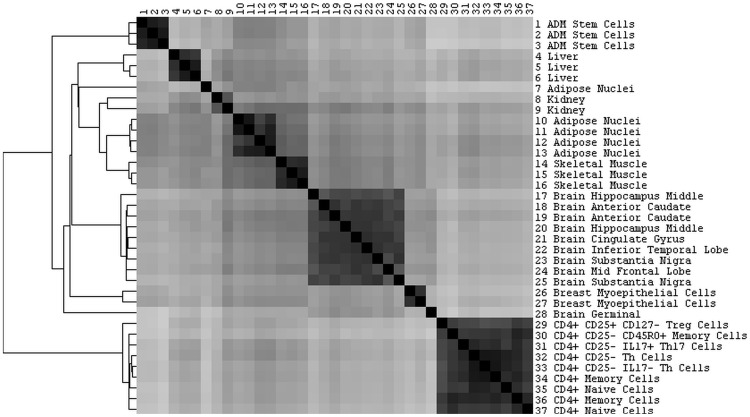
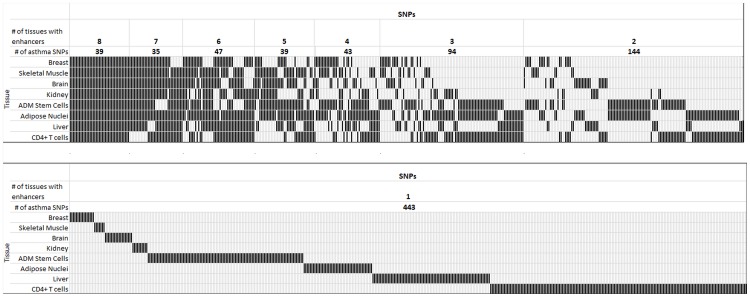
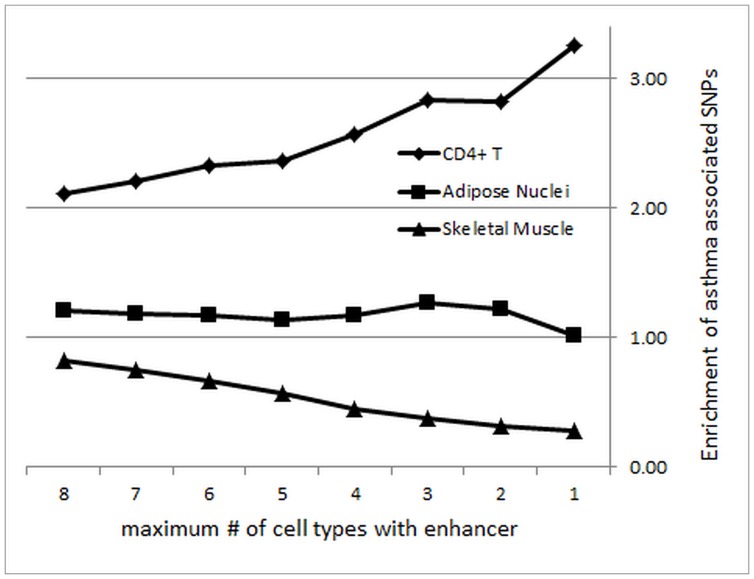
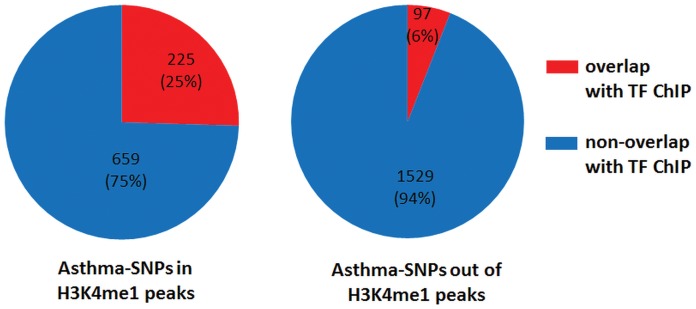
Genome-wide association studies (GWASs) identify single nucleotide polymorphisms (SNPs) that are enriched in individuals suffering from a given disease. Most disease-associated SNPs fall into non-coding regions, so that it is not straightforward to infer phenotype or function; moreover, many SNPs are in tight genetic linkage, so that a SNP identified as associated with a particular disease may not itself be causal, but rather signify the presence of a linked SNP that is functionally relevant to disease pathogenesis. Here, we present an analysis method that takes advantage of the recent rapid accumulation of epigenomics data to address these problems for some SNPs. Using asthma as a prototypic example; we show that non-coding disease-associated SNPs are enriched in genomic regions that function as regulators of transcription, such as enhancers and promoters. Identifying enhancers based on the presence of the histone modification marks such as H3K4me1 in different cell types, we show that the location of enhancers is highly cell-type specific. We use these findings to predict which SNPs are likely to be directly contributing to disease based on their presence in regulatory regions, and in which cell types their effect is expected to be detectable. Moreover, we can also predict which cell types contribute to a disease based on overlap of the disease-associated SNPs with the locations of enhancers present in a given cell type. Finally, we suggest that it will be possible to re-analyze GWAS studies with much higher power by limiting the SNPs considered to those in coding or regulatory regions of cell types relevant to a given disease.
全基因组关联研究 (GWAS) 确定了在患有特定疾病的个体中富集的单核苷酸多态性 (SNPs)。大多数与疾病相关的 SNPs 位于非编码区域,因此很难推断表型或功能;此外,许多 SNPs 紧密连锁,因此被确定与特定疾病相关的 SNP 本身可能不是因果关系,而是标志着与疾病发病机制相关的连锁 SNP 的存在。在这里,我们提出了一种分析方法,利用最近快速积累的表观基因组学数据来解决这些问题。以哮喘为例;我们表明,非编码疾病相关 SNPs 在作为转录调节剂的基因组区域中富集,例如增强子和启动子。基于不同细胞类型中存在的组蛋白修饰标记(如 H3K4me1)来识别增强子,我们表明增强子的位置高度细胞类型特异性。我们利用这些发现,根据它们在调节区域中的存在以及在哪些细胞类型中可以检测到其效应,预测哪些 SNPs 可能直接导致疾病。此外,我们还可以根据疾病相关 SNPs 与特定细胞类型中存在的增强子位置的重叠来预测哪些细胞类型与疾病有关。最后,我们建议通过将考虑的 SNPs 限制为与特定疾病相关的细胞类型的编码或调节区域,可以对 GWAS 研究进行更具说服力的重新分析。