Computational Biology and Machine Learning Laboratory, Center for Cancer Research and Cell Biology, School of Medicine, Dentistry and Biomedical Sciences, Queen's University Belfast, Belfast, 97 Lisburn Road, Belfast BT9 7BL, UK.
Nucleic Acids Res. 2013 Apr;41(7):e82. doi: 10.1093/nar/gkt054. Epub 2013 Feb 6.
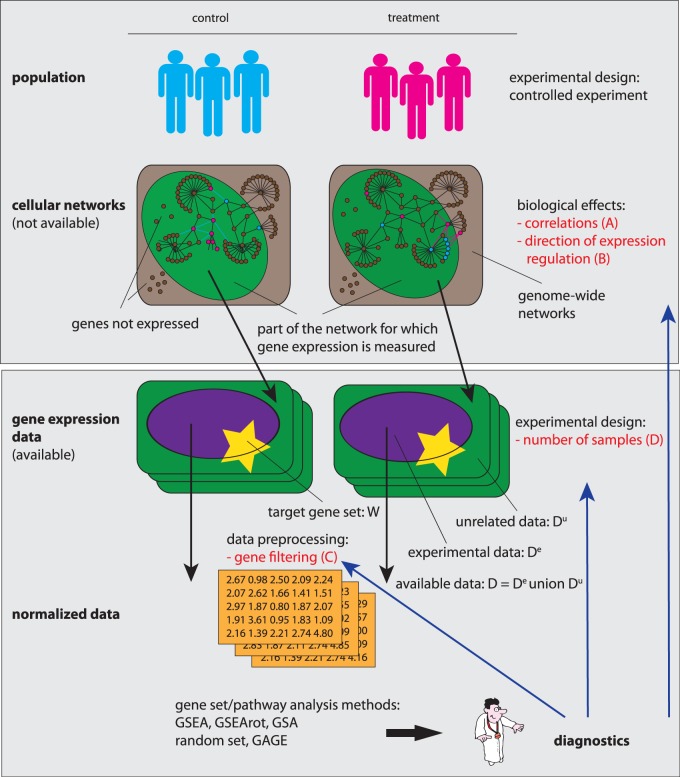
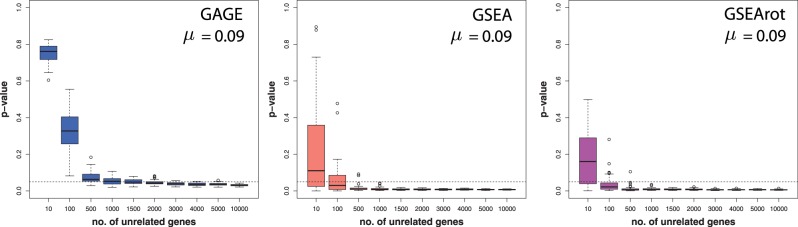
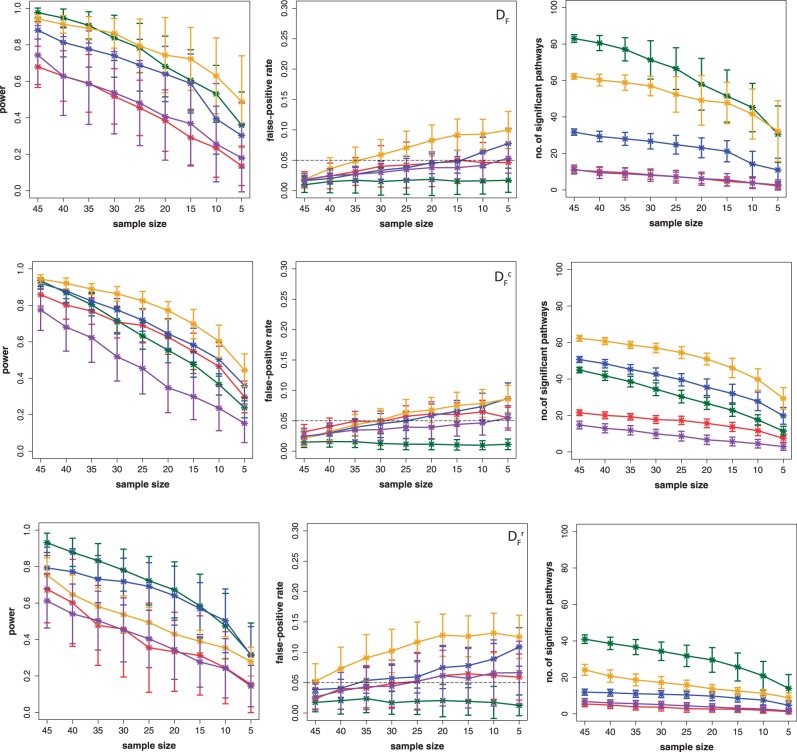
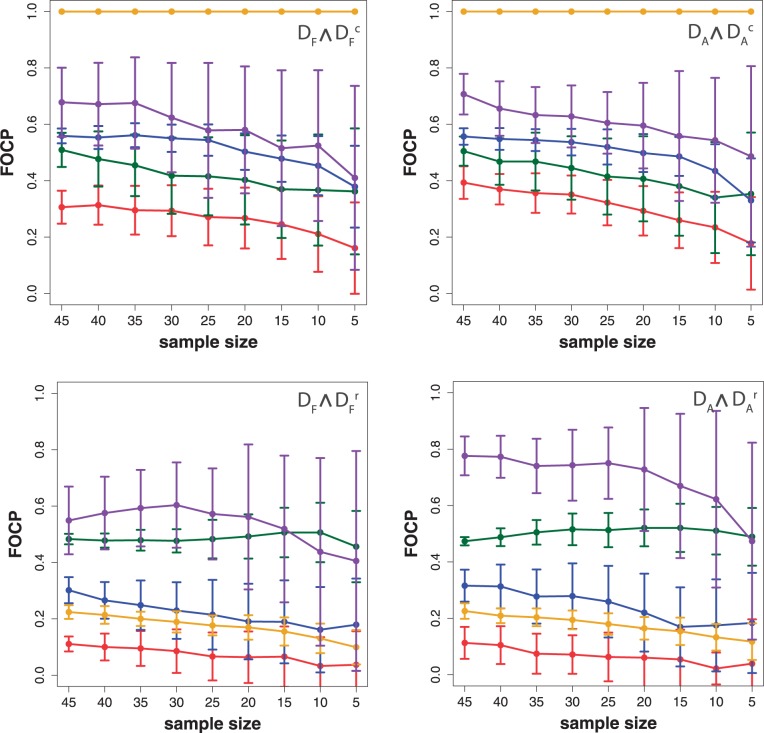
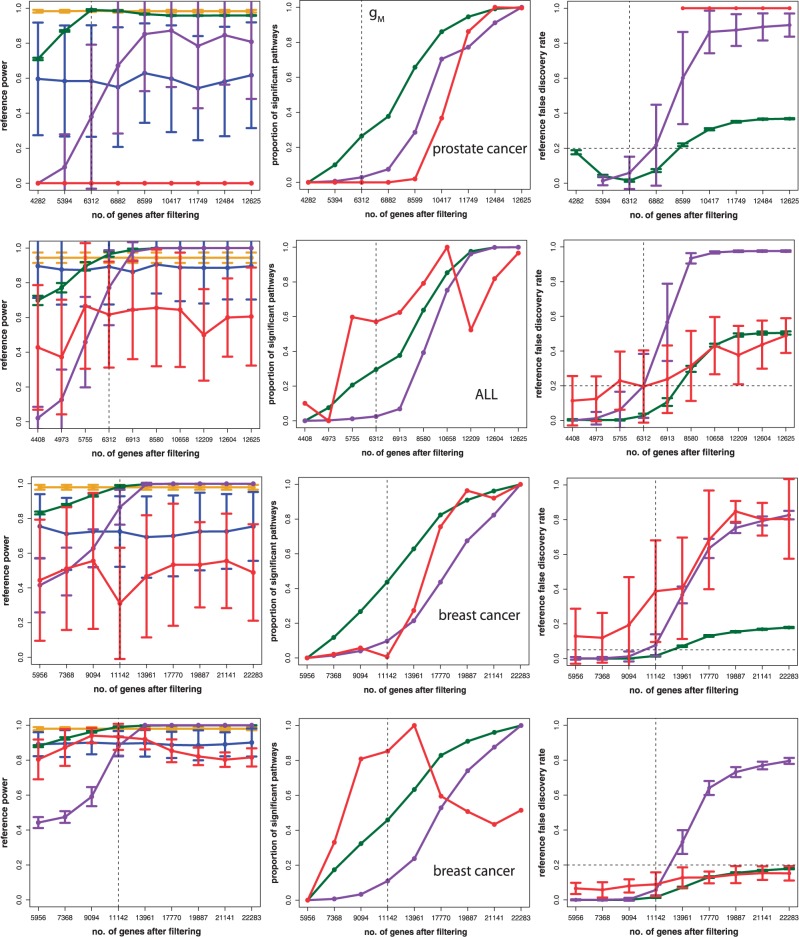
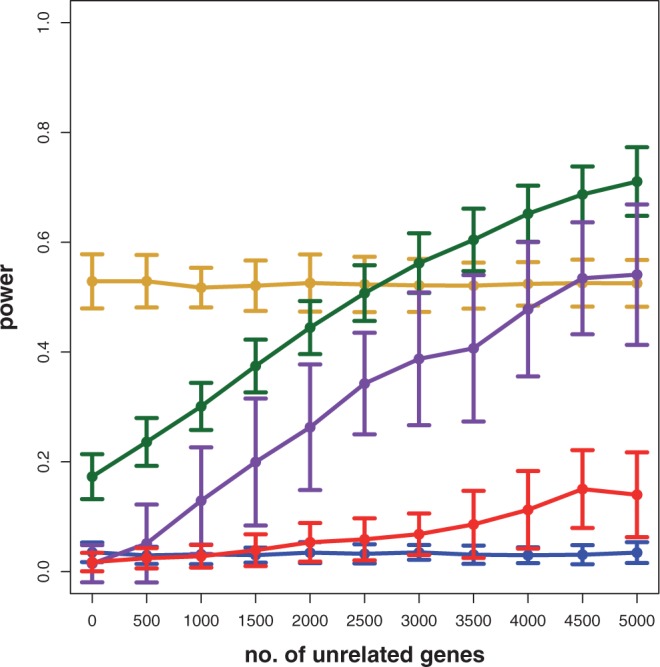
In this article, we focus on the analysis of competitive gene set methods for detecting the statistical significance of pathways from gene expression data. Our main result is to demonstrate that some of the most frequently used gene set methods, GSEA, GSEArot and GAGE, are severely influenced by the filtering of the data in a way that such an analysis is no longer reconcilable with the principles of statistical inference, rendering the obtained results in the worst case inexpressive. A possible consequence of this is that these methods can increase their power by the addition of unrelated data and noise. Our results are obtained within a bootstrapping framework that allows a rigorous assessment of the robustness of results and enables power estimates. Our results indicate that when using competitive gene set methods, it is imperative to apply a stringent gene filtering criterion. However, even when genes are filtered appropriately, for gene expression data from chips that do not provide a genome-scale coverage of the expression values of all mRNAs, this is not enough for GSEA, GSEArot and GAGE to ensure the statistical soundness of the applied procedure. For this reason, for biomedical and clinical studies, we strongly advice not to use GSEA, GSEArot and GAGE for such data sets.
在本文中,我们专注于分析竞争基因集方法,以检测基因表达数据中途径的统计学意义。我们的主要结果是证明,一些最常用的基因集方法,GSEA、GSEArot 和 GAGE,受到数据过滤的严重影响,以至于这种分析不再与统计推断的原则一致,使得在最坏的情况下得到的结果没有表达力。这种情况的一个可能后果是,这些方法可以通过添加不相关的数据和噪声来增加其功效。我们的结果是在一个自举框架内获得的,该框架允许对结果的稳健性进行严格评估,并能够进行功效估计。我们的结果表明,在使用竞争基因集方法时,必须应用严格的基因过滤标准。然而,即使对芯片的基因表达数据进行适当的基因过滤,对于不能提供所有 mRNA 表达值的全基因组覆盖的基因表达数据,对于 GSEA、GSEArot 和 GAGE 来说,这不足以确保所应用程序的统计合理性。因此,对于生物医学和临床研究,我们强烈建议不要在这种数据集上使用 GSEA、GSEArot 和 GAGE。