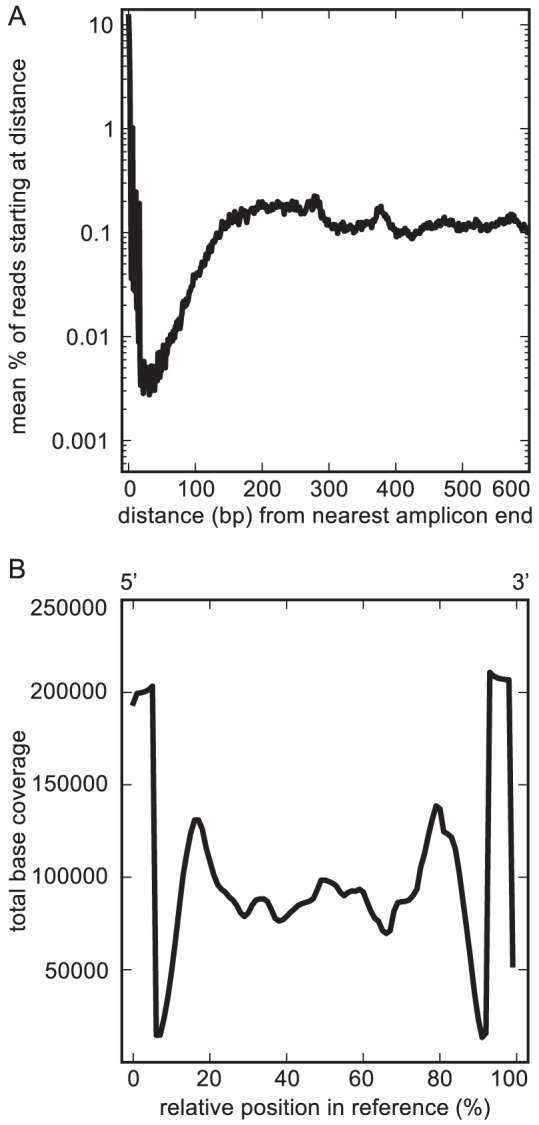
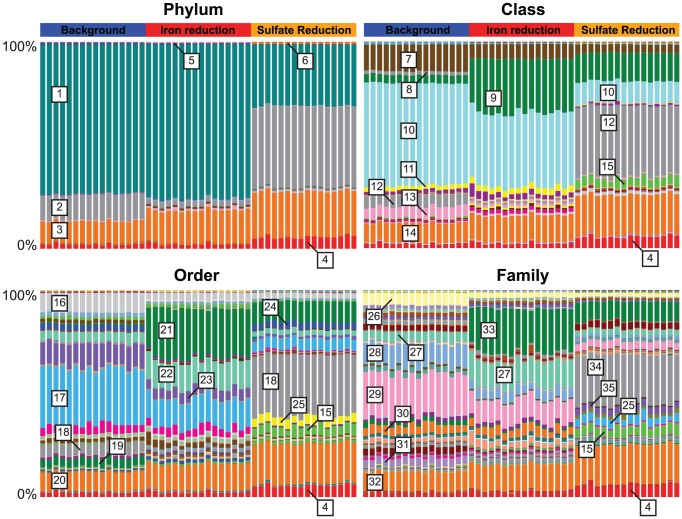
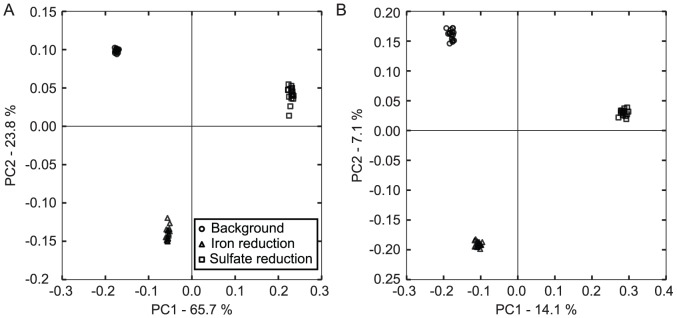
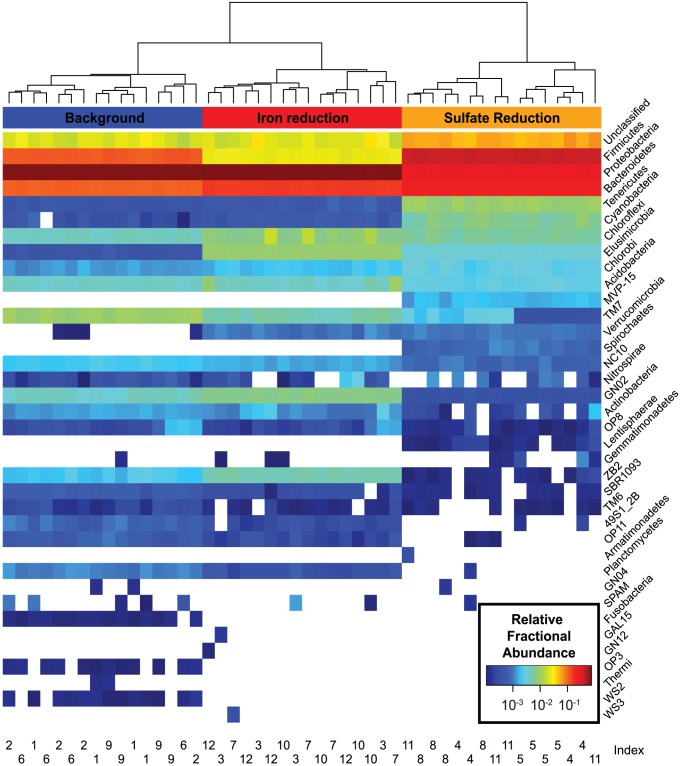
Department of Earth and Planetary Science, University of California, Berkeley, California, United States of America.
PLoS One. 2013;8(2):e56018. doi: 10.1371/journal.pone.0056018. Epub 2013 Feb 6.
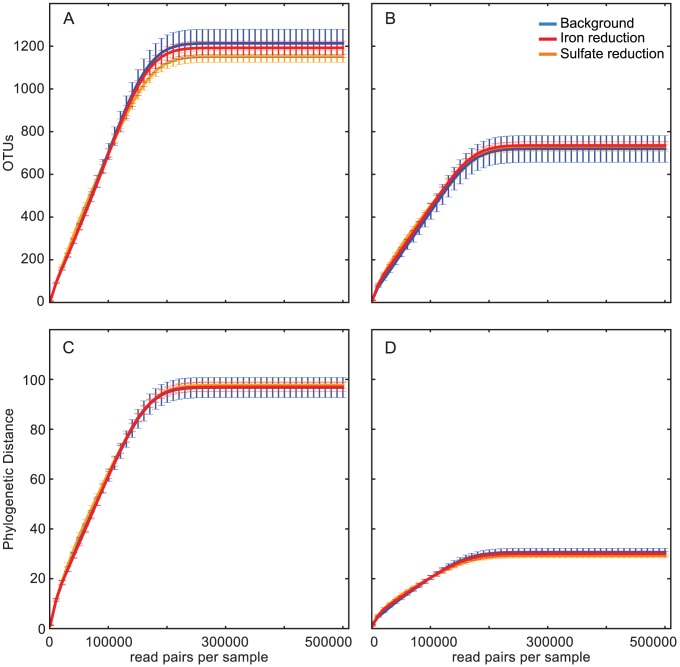
In microbial ecology, a fundamental question relates to how community diversity and composition change in response to perturbation. Most studies have had limited ability to deeply sample community structure (e.g. Sanger-sequenced 16S rRNA libraries), or have had limited taxonomic resolution (e.g. studies based on 16S rRNA hypervariable region sequencing). Here, we combine the higher taxonomic resolution of near-full-length 16S rRNA gene amplicons with the economics and sensitivity of short-read sequencing to assay the abundance and identity of organisms that represent as little as 0.01% of sediment bacterial communities. We used a new version of EMIRGE optimized for large data size to reconstruct near-full-length 16S rRNA genes from amplicons sheared and sequenced with Illumina technology. The approach allowed us to differentiate the community composition among samples acquired before perturbation, after acetate amendment shifted the predominant metabolism to iron reduction, and once sulfate reduction began. Results were highly reproducible across technical replicates, and identified specific taxa that responded to the perturbation. All samples contain very high alpha diversity and abundant organisms from phyla without cultivated representatives. Surprisingly, at the time points measured, there was no strong loss of evenness, despite the selective pressure of acetate amendment and change in the terminal electron accepting process. However, community membership was altered significantly. The method allows for sensitive, accurate profiling of the "long tail" of low abundance organisms that exist in many microbial communities, and can resolve population dynamics in response to environmental change.
在微生物生态学中,一个基本的问题是群落多样性和组成如何响应扰动而变化。大多数研究都只能有限地深入采样群落结构(例如,Sanger 测序的 16S rRNA 文库),或者只能有限地解析分类(例如,基于 16S rRNA 高变区测序的研究)。在这里,我们将 16S rRNA 基因全长扩增子的更高分类分辨率与短读测序的经济性和敏感性相结合,检测了仅占沉积物细菌群落 0.01%的生物体的丰度和身份。我们使用了经过优化可处理大数据量的新版本 EMIRGE,从用 Illumina 技术剪切和测序的扩增子中重建近全长 16S rRNA 基因。该方法使我们能够区分在扰动前、在乙酸盐添加后代谢主要转变为铁还原、以及硫酸盐还原开始时获得的样品之间的群落组成。结果在技术重复中具有高度的可重复性,并鉴定了对扰动有响应的特定分类群。所有样品都包含非常高的 alpha 多样性和丰富的无培养代表的门生物。令人惊讶的是,在测量的时间点上,尽管有乙酸盐添加的选择压力和末端电子接受过程的改变,均匀度并没有强烈丧失。然而,群落成员发生了显著的改变。该方法可以敏感、准确地描绘存在于许多微生物群落中的低丰度生物体的“长尾”,并能够解析对环境变化的种群动态。