Lehrstuhl fuer Tierzucht, Technische Universitaet Muenchen, 85354, Freising, Germany.
Genet Sel Evol. 2013 Feb 13;45(1):3. doi: 10.1186/1297-9686-45-3.
Currently, genome-wide evaluation of cattle populations is based on SNP-genotyping using ~ 54,000 SNP. Increasing the number of markers might improve genomic predictions and power of genome-wide association studies. Imputation of genotypes makes it possible to extrapolate genotypes from lower to higher density arrays based on a representative reference sample for which genotypes are obtained at higher density.
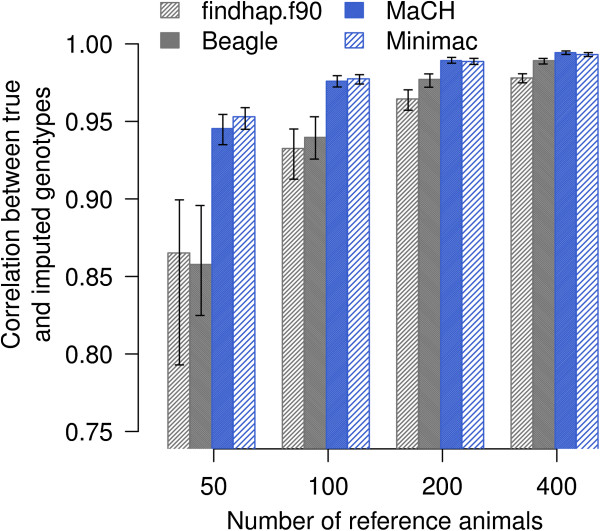
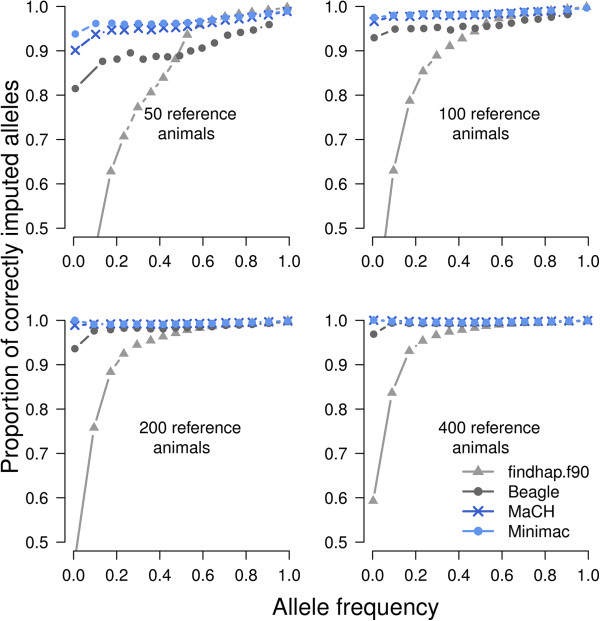
Genotypes using 639 214 SNP were available for 797 bulls of the Fleckvieh cattle breed. The data set was divided into a reference and a validation population. Genotypes for all SNP except those included in the BovineSNP50 Bead chip were masked and subsequently imputed for animals of the validation population. Imputation of genotypes was performed with Beagle, findhap.f90, MaCH and Minimac. The accuracy of the imputed genotypes was assessed for four different scenarios including 50, 100, 200 and 400 animals as reference population. The reference animals were selected to account for 78.03%, 89.21%, 97.47% and > 99% of the gene pool of the genotyped population, respectively.
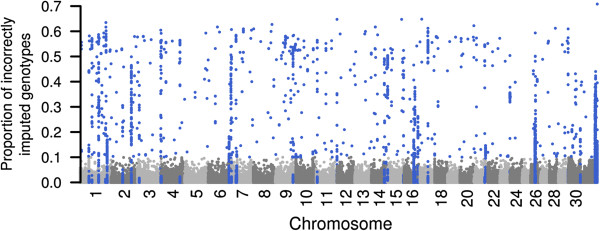
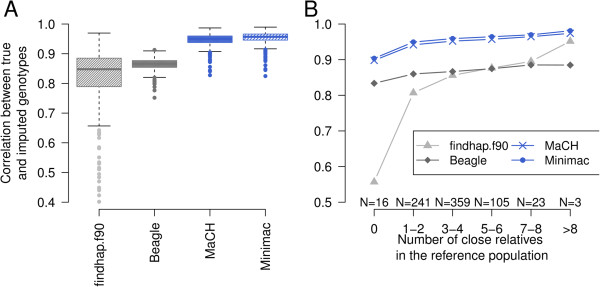
Imputation accuracy increased as the number of animals and relatives in the reference population increased. Population-based algorithms provided highly reliable imputation of genotypes, even for scenarios with 50 and 100 reference animals only. Using MaCH and Minimac, the correlation between true and imputed genotypes was > 0.975 with 100 reference animals only. Pre-phasing the genotypes of both the reference and validation populations not only provided highly accurate imputed genotypes but was also computationally efficient. Genome-wide analysis of imputation accuracy led to the identification of many misplaced SNP.
Genotyping key animals at high density and subsequent population-based genotype imputation yield high imputation accuracy. Pre-phasing the genotypes of the reference and validation populations is computationally efficient and results in high imputation accuracy, even when the reference population is small.
目前,对牛群的全基因组评估是基于使用约 54000 个 SNP 的 SNP 基因分型。增加标记数量可能会提高基因组预测和全基因组关联研究的能力。基因型的推断使得基于具有更高密度基因型的代表性参考样本,从较低密度的阵列推断基因型成为可能。
为弗莱维赫牛种的 797 头公牛提供了使用 639214 个 SNP 的基因型。数据集分为参考群体和验证群体。除包含在 BovineSNP50 Bead 芯片中的 SNP 之外,所有 SNP 的基因型都被屏蔽,随后对验证群体中的动物进行了基因型推断。使用 Beagle、findhap.f90、MaCH 和 Minimac 进行基因型推断。对于四种不同的情况,评估了推断基因型的准确性,包括将 50、100、200 和 400 头动物作为参考群体。参考动物的选择分别考虑了遗传群体的 78.03%、89.21%、97.47%和 > 99%。
随着参考群体中动物数量和亲属数量的增加,推断准确性增加。基于群体的算法提供了高度可靠的基因型推断,即使在只有 50 和 100 个参考动物的情况下也是如此。使用 MaCH 和 Minimac,仅使用 100 个参考动物,真实基因型和推断基因型之间的相关性 > 0.975。对参考和验证群体的基因型进行预相位不仅提供了高度准确的推断基因型,而且计算效率也很高。全基因组推断准确性分析导致了许多错位 SNP 的识别。
对高密度关键动物进行基因分型,并随后进行基于群体的基因型推断,可以获得高度准确的推断。对参考和验证群体的基因型进行预相位计算效率高,即使参考群体较小,也能获得高度准确的推断。