Boyle Amanda J, Roddick Leigh Ann, Bhakta Varsha, Lambourne Melissa D, Junop Murray S, Liaw Patricia C, Weitz Jeffrey I, Sheffield William P
BMC Biochem. 2013 Mar 7;14:6. doi: 10.1186/1471-2091-14-6.
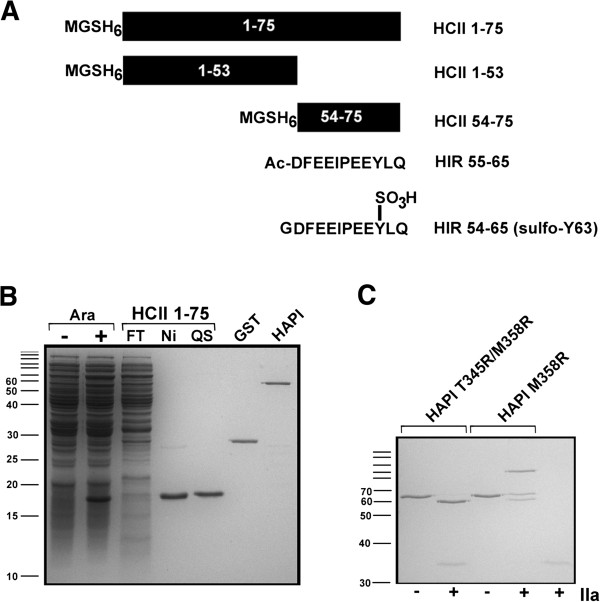
Heparin cofactor II (HCII) is a circulating protease inhibitor, one which contains an N-terminal acidic extension (HCII 1-75) unique within the serpin superfamily. Deletion of HCII 1-75 greatly reduces the ability of glycosaminoglycans (GAGs) to accelerate the inhibition of thrombin, and abrogates HCII binding to thrombin exosite 1. While a minor portion of HCII 1-75 can be visualized in a crystallized HCII-thrombin S195A complex, the role of the rest of the extension is not well understood and the affinity of the HCII 1-75 interaction has not been quantitatively characterized. To address these issues, we expressed HCII 1-75 as a small, N-terminally hexahistidine-tagged polypeptide in E. coli.
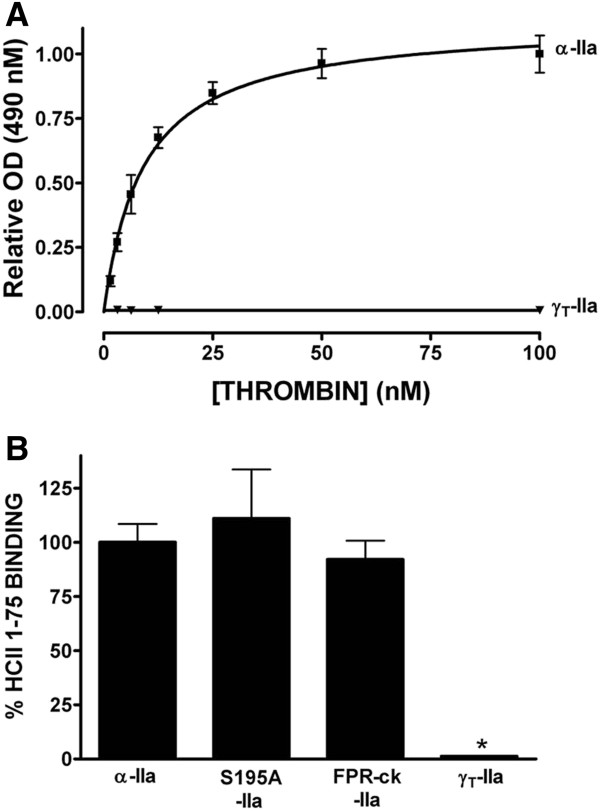
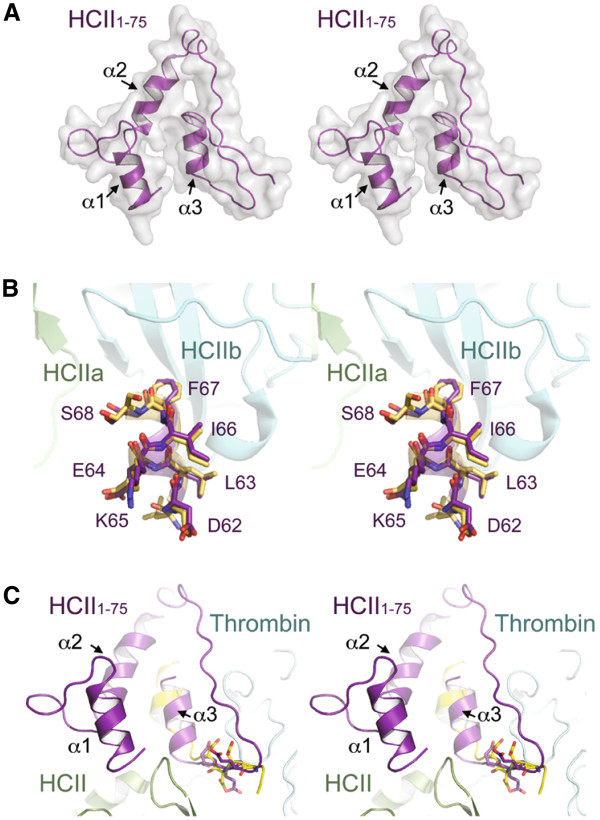
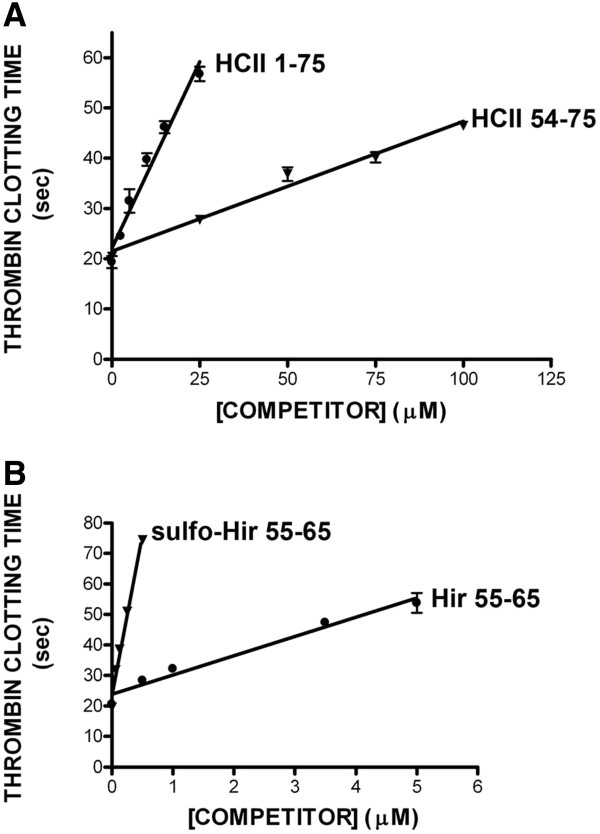
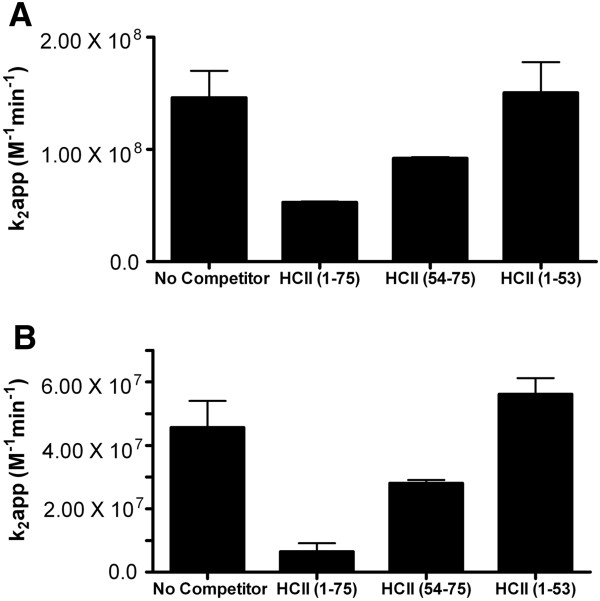
Immobilized purified HCII 1-75 bound active α-thrombin and active-site inhibited FPR-ck- or S195A-thrombin, but not exosite-1-disrupted γT-thrombin, in microtiter plate assays. Biotinylated HCII 1-75 immobilized on streptavidin chips bound α-thrombin and FPR-ck-thrombin with similar KD values of 330-340 nM. HCII 1-75 competed thrombin binding to chip-immobilized HCII 1-75 more effectively than HCII 54-75 but less effectively than the C-terminal dodecapeptide of hirudin (mean Ki values of 2.6, 8.5, and 0.29 μM, respectively). This superiority over HCII 54-75 was also demonstrated in plasma clotting assays and in competing the heparin-catalysed inhibition of thrombin by plasma-derived HCII; HCII 1-53 had no effect in either assay. Molecular modelling of HCII 1-75 correctly predicted those portions of the acidic extension that had been previously visualized in crystal structures, and suggested that an α-helix found between residues 26 and 36 stabilizes one found between residues 61-67. The latter region has been previously shown by deletion mutagenesis and crystallography to play a crucial role in the binding of HCII to thrombin exosite 1.
Assuming that the KD value for HCII 1-75 of 330-340 nM faithfully predicts that of this region in intact HCII, and that 1-75 binding to exosite 1 is GAG-dependent, our results support a model in which thrombin first binds to GAGs, followed by HCII addition to the ternary complex and release of HCII 1-75 for exosite 1 binding and serpin mechanism inhibition. They further suggest that, in isolated or transferred form, the entire HCII 1-75 region is required to ensure maximal binding of thrombin exosite 1.
肝素辅因子II(HCII)是一种循环蛋白酶抑制剂,其在丝氨酸蛋白酶抑制剂超家族中含有独特的N端酸性延伸序列(HCII 1 - 75)。删除HCII 1 - 75会大大降低糖胺聚糖(GAGs)加速凝血酶抑制的能力,并消除HCII与凝血酶外位点1的结合。虽然在结晶的HCII - 凝血酶S195A复合物中可以看到一小部分HCII 1 - 75,但该延伸序列其余部分的作用尚不清楚,且HCII 1 - 75相互作用的亲和力尚未得到定量表征。为了解决这些问题,我们在大肠杆菌中表达了HCII 1 - 75,它是一种N端带有六聚组氨酸标签的小多肽。
在微量滴定板试验中,固定化的纯化HCII 1 - 75能结合活性α-凝血酶和活性位点被抑制的FPR - ck - 或S195A - 凝血酶,但不能结合外位点1被破坏的γT - 凝血酶。固定在链霉亲和素芯片上的生物素化HCII 1 - 75以相似的330 - 340 nM的KD值结合α-凝血酶和FPR - ck - 凝血酶。HCII 1 - 75比HCII 54 - 75更有效地竞争凝血酶与芯片固定的HCII 1 - 75的结合,但比水蛭素的C端十二肽效率低(平均Ki值分别为2.6、8.5和0.29 μM)。在血浆凝血试验以及竞争血浆来源的HCII对肝素催化的凝血酶抑制作用中,HCII 1 - 75相对于HCII 54 - 75的这种优势也得到了证明;HCII 1 - 53在这两种试验中均无作用。HCII 1 - 75的分子建模正确预测了先前在晶体结构中看到的酸性延伸序列的那些部分,并表明在26至36位残基之间发现的α-螺旋稳定了61 - 67位残基之间的α-螺旋。先前通过缺失诱变和晶体学研究表明,后一个区域在HCII与凝血酶外位点1的结合中起关键作用。
假设HCII 1 - 75的330 - 340 nM的KD值如实地预测了完整HCII中该区域的值,并且1 - 75与外位点1的结合是GAG依赖性的,我们的结果支持一种模型,即凝血酶首先与GAGs结合,随后HCII添加到三元复合物中,并释放HCII 1 - 75用于外位点1的结合和丝氨酸蛋白酶抑制剂机制抑制。它们进一步表明,以分离或转移形式存在时,需要整个HCII 1 - 75区域来确保凝血酶外位点1的最大结合。