Department of Mathematics, University of Bristol, Bristol, United Kingdom.
PLoS Genet. 2012 Jan;8(1):e1002453. doi: 10.1371/journal.pgen.1002453. Epub 2012 Jan 26.
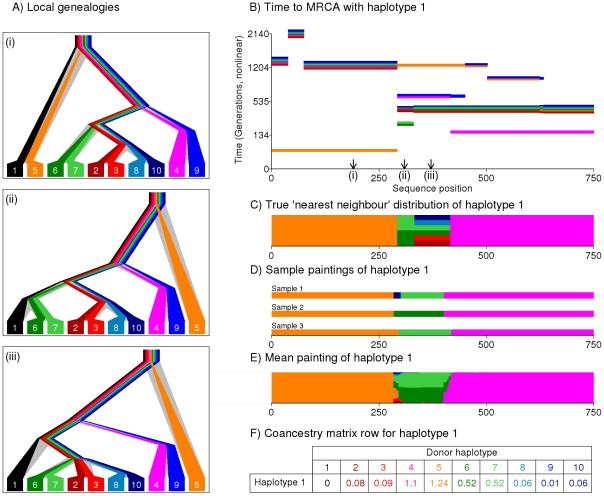
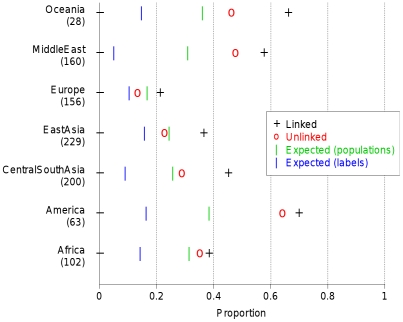
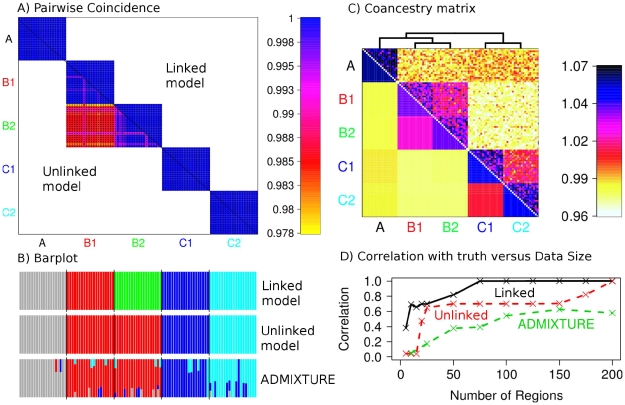
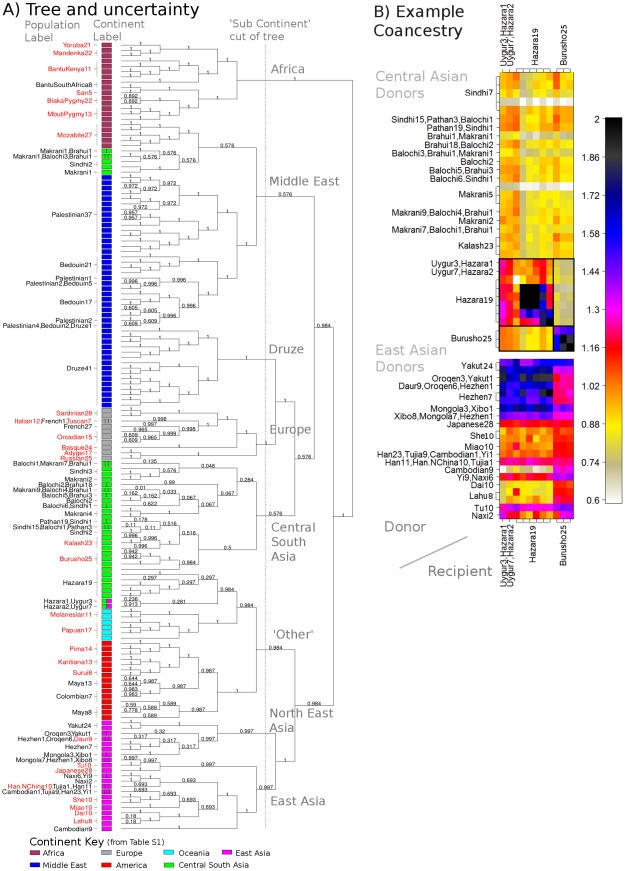
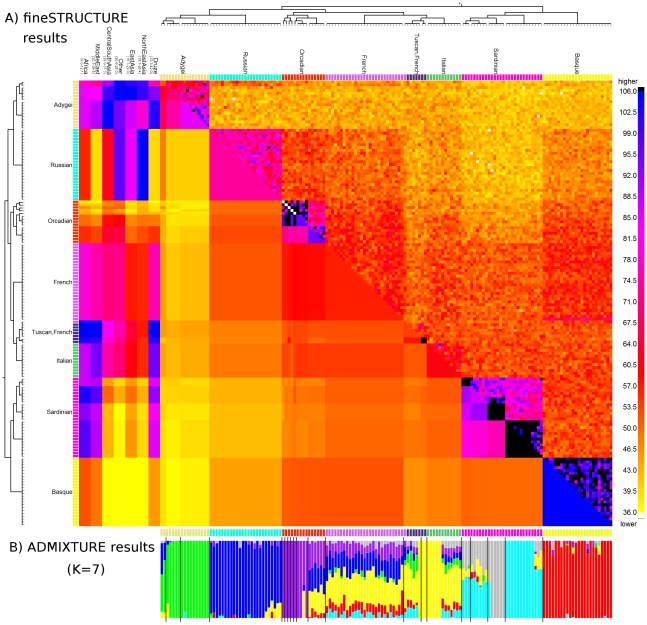
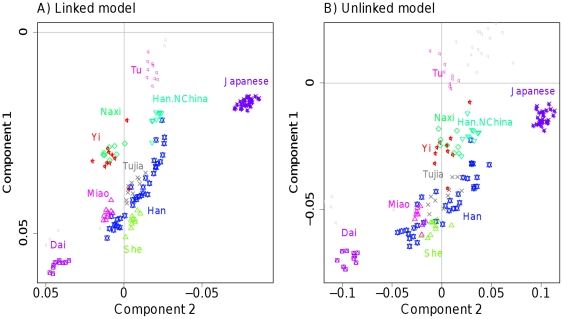
The advent of genome-wide dense variation data provides an opportunity to investigate ancestry in unprecedented detail, but presents new statistical challenges. We propose a novel inference framework that aims to efficiently capture information on population structure provided by patterns of haplotype similarity. Each individual in a sample is considered in turn as a recipient, whose chromosomes are reconstructed using chunks of DNA donated by the other individuals. Results of this "chromosome painting" can be summarized as a "coancestry matrix," which directly reveals key information about ancestral relationships among individuals. If markers are viewed as independent, we show that this matrix almost completely captures the information used by both standard Principal Components Analysis (PCA) and model-based approaches such as STRUCTURE in a unified manner. Furthermore, when markers are in linkage disequilibrium, the matrix combines information across successive markers to increase the ability to discern fine-scale population structure using PCA. In parallel, we have developed an efficient model-based approach to identify discrete populations using this matrix, which offers advantages over PCA in terms of interpretability and over existing clustering algorithms in terms of speed, number of separable populations, and sensitivity to subtle population structure. We analyse Human Genome Diversity Panel data for 938 individuals and 641,000 markers, and we identify 226 populations reflecting differences on continental, regional, local, and family scales. We present multiple lines of evidence that, while many methods capture similar information among strongly differentiated groups, more subtle population structure in human populations is consistently present at a much finer level than currently available geographic labels and is only captured by the haplotype-based approach. The software used for this article, ChromoPainter and fineSTRUCTURE, is available from http://www.paintmychromosomes.com/.
全基因组高密度变异数据的出现为我们提供了一个前所未有的机会来详细研究人类的祖先,然而也带来了新的统计挑战。我们提出了一种新的推断框架,旨在有效地捕捉由单倍型相似性模式提供的群体结构信息。样本中的每个个体依次被视为接受者,其染色体使用其他个体捐献的 DNA 块进行重建。这种“染色体绘画”的结果可以总结为“共祖矩阵”,它直接揭示了个体之间祖先关系的关键信息。如果将标记视为独立的,我们将表明,该矩阵几乎以统一的方式完全捕获了标准主成分分析(PCA)和基于模型的方法(如 STRUCTURE)所使用的信息。此外,当标记处于连锁不平衡时,该矩阵将跨连续标记的信息组合起来,以增加使用 PCA 识别细微群体结构的能力。与此同时,我们开发了一种有效的基于模型的方法来使用该矩阵识别离散群体,该方法在可解释性方面优于 PCA,在速度、可分离群体数量和对细微群体结构的敏感性方面优于现有的聚类算法。我们分析了 938 个人和 641000 个标记的人类基因组多样性面板数据,并识别出 226 个反映大陆、区域、局部和家庭尺度差异的群体。我们提供了多条证据表明,虽然许多方法在高度分化的群体中捕捉到了相似的信息,但人类群体中更细微的群体结构始终以比当前可用的地理标签更精细的水平存在,并且只能通过基于单倍型的方法来捕捉。本文使用的软件,ChromoPainter 和 fineSTRUCTURE,可从 http://www.paintmychromosomes.com/ 获取。