Department of Computer Science, Princeton University, Princeton, New Jersey, USA.
PLoS Comput Biol. 2013;9(3):e1002957. doi: 10.1371/journal.pcbi.1002957. Epub 2013 Mar 14.
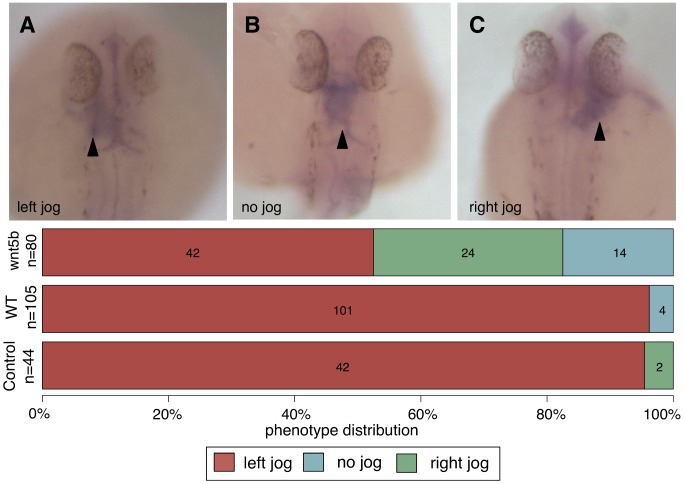
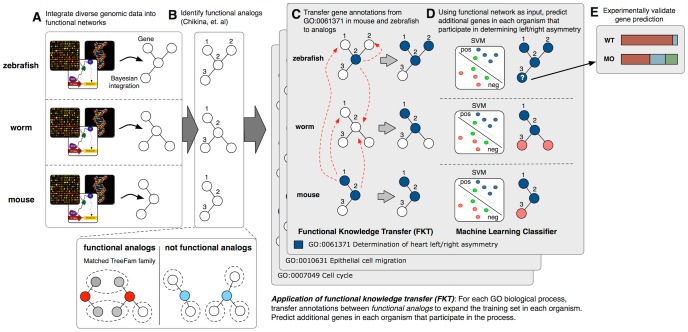
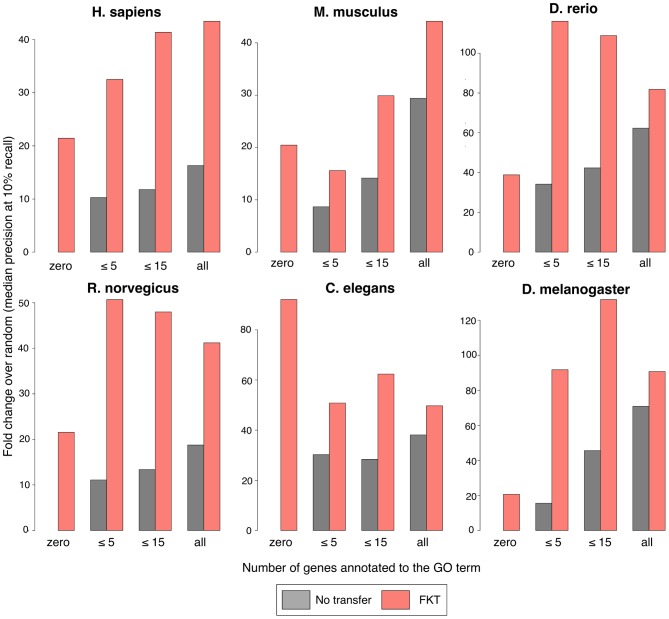
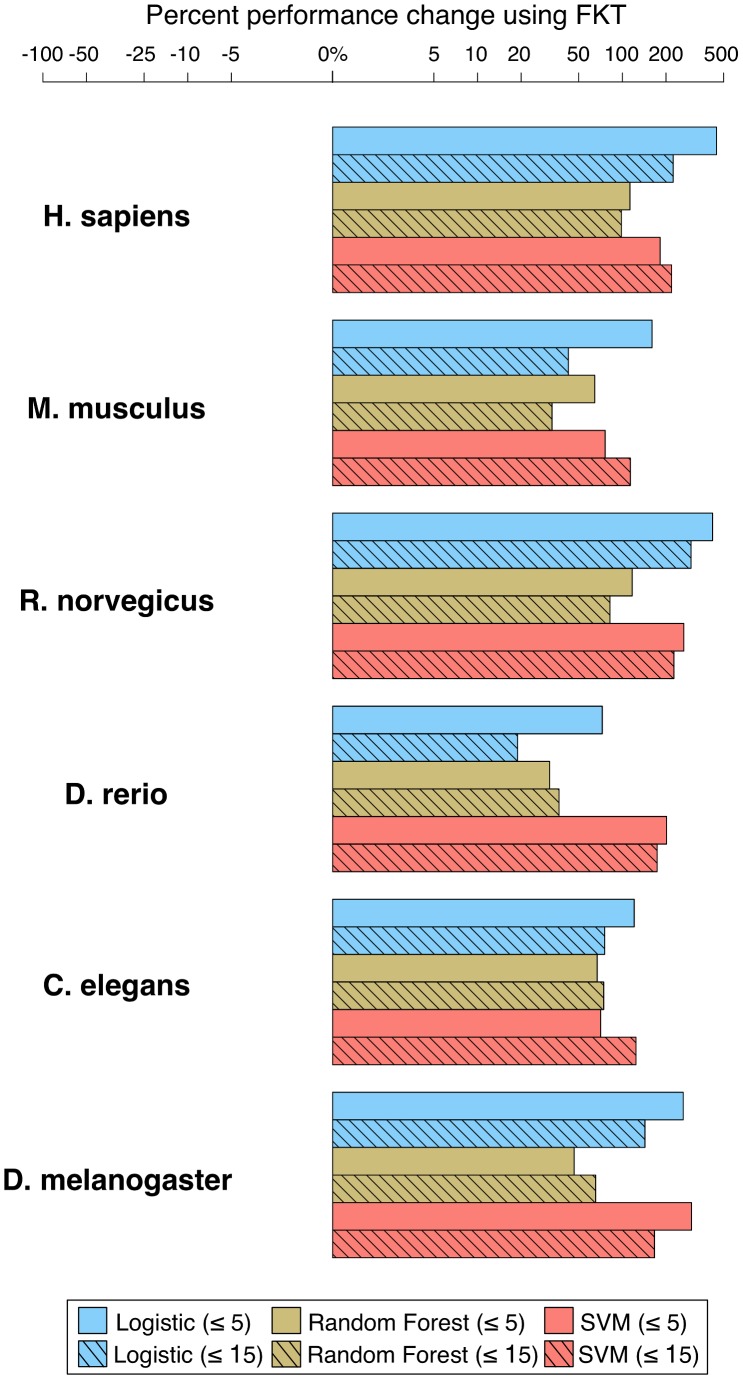
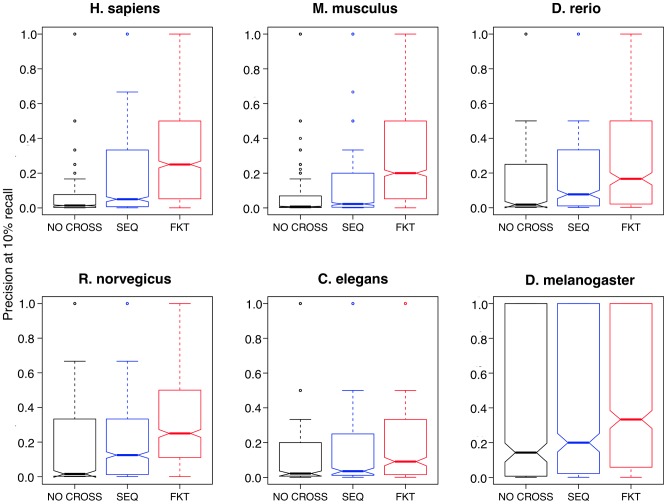
A key challenge in genetics is identifying the functional roles of genes in pathways. Numerous functional genomics techniques (e.g. machine learning) that predict protein function have been developed to address this question. These methods generally build from existing annotations of genes to pathways and thus are often unable to identify additional genes participating in processes that are not already well studied. Many of these processes are well studied in some organism, but not necessarily in an investigator's organism of interest. Sequence-based search methods (e.g. BLAST) have been used to transfer such annotation information between organisms. We demonstrate that functional genomics can complement traditional sequence similarity to improve the transfer of gene annotations between organisms. Our method transfers annotations only when functionally appropriate as determined by genomic data and can be used with any prediction algorithm to combine transferred gene function knowledge with organism-specific high-throughput data to enable accurate function prediction. We show that diverse state-of-art machine learning algorithms leveraging functional knowledge transfer (FKT) dramatically improve their accuracy in predicting gene-pathway membership, particularly for processes with little experimental knowledge in an organism. We also show that our method compares favorably to annotation transfer by sequence similarity. Next, we deploy FKT with state-of-the-art SVM classifier to predict novel genes to 11,000 biological processes across six diverse organisms and expand the coverage of accurate function predictions to processes that are often ignored because of a dearth of annotated genes in an organism. Finally, we perform in vivo experimental investigation in Danio rerio and confirm the regulatory role of our top predicted novel gene, wnt5b, in leftward cell migration during heart development. FKT is immediately applicable to many bioinformatics techniques and will help biologists systematically integrate prior knowledge from diverse systems to direct targeted experiments in their organism of study.
在遗传学中,一个关键的挑战是确定基因在途径中的功能作用。已经开发了许多预测蛋白质功能的功能基因组学技术(例如机器学习)来解决这个问题。这些方法通常是基于基因到途径的现有注释构建的,因此往往无法识别参与尚未充分研究的过程的其他基因。这些过程中的许多在某些生物体中得到了很好的研究,但在研究人员感兴趣的生物体中不一定得到了很好的研究。基于序列的搜索方法(例如 BLAST)已被用于在生物体之间转移这种注释信息。我们证明功能基因组学可以补充传统的序列相似性,以提高基因注释在生物体之间的转移。我们的方法仅在功能上合适时才会转移注释,这是由基因组数据确定的,并且可以与任何预测算法一起使用,将转移的基因功能知识与特定于生物体的高通量数据相结合,以实现准确的功能预测。我们表明,利用功能知识转移(FKT)的各种最先进的机器学习算法可以显著提高它们预测基因途径成员的准确性,特别是对于在生物体中实验知识很少的过程。我们还表明,我们的方法与基于序列相似性的注释转移相比具有优势。接下来,我们使用 FKT 和最先进的 SVM 分类器来预测 6 个不同生物体的 11000 个生物学过程中的新基因,并将准确功能预测的覆盖范围扩展到经常由于生物体中注释基因缺乏而被忽略的过程。最后,我们在 Danio rerio 中进行了体内实验研究,并证实了我们预测的新基因 wnt5b 在心脏发育过程中向左细胞迁移的调节作用。FKT 立即适用于许多生物信息学技术,并将帮助生物学家系统地整合来自不同系统的先验知识,以指导其研究生物体中的靶向实验。