Colombian Center for Genomics and Bioinformatics of Extreme Environments Gebix, Bogota, Colombia.
PLoS One. 2013;8(3):e59488. doi: 10.1371/journal.pone.0059488. Epub 2013 Mar 25.
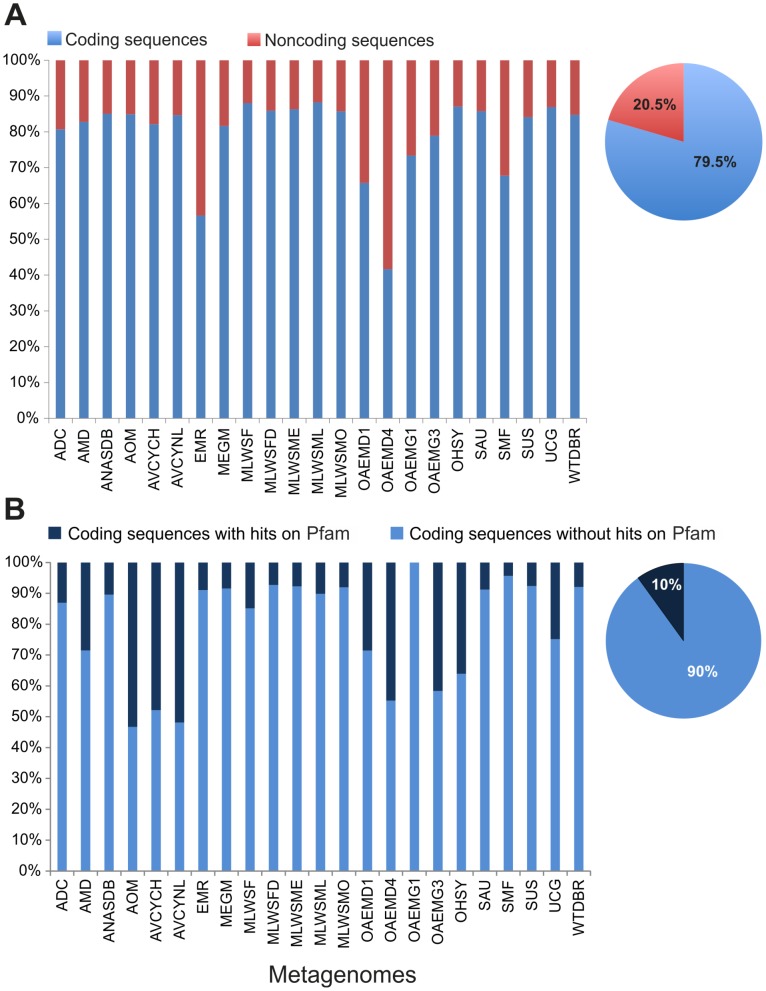
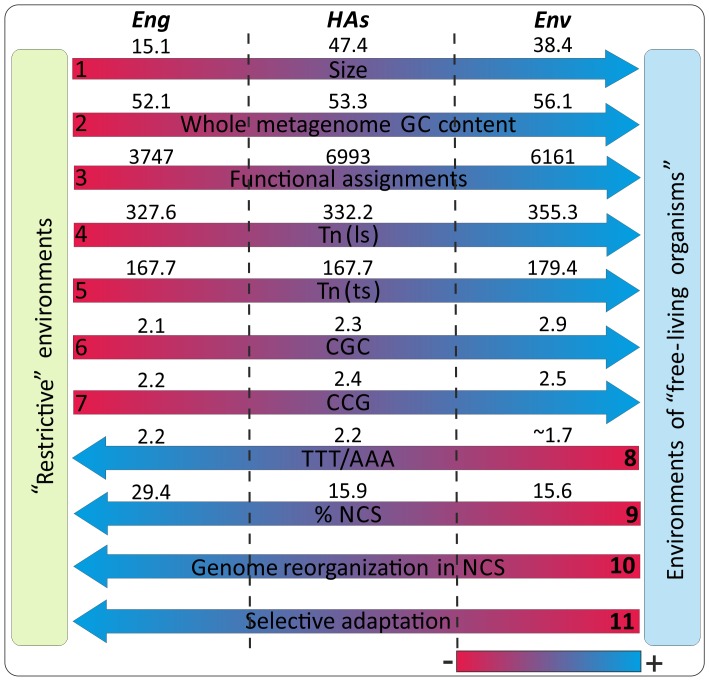
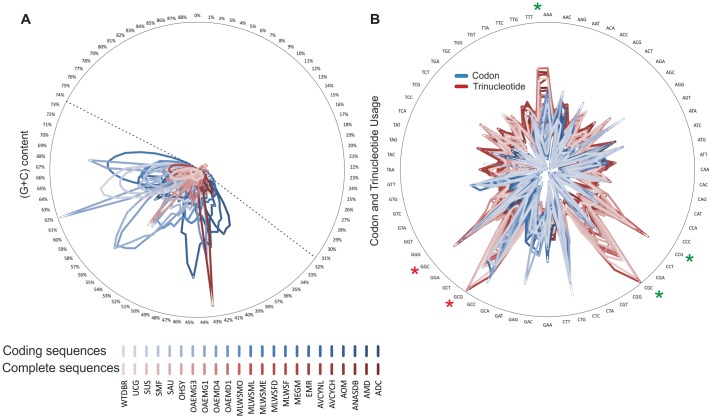
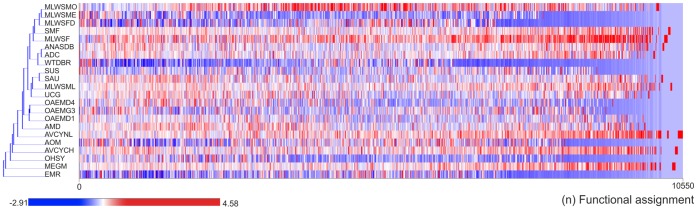
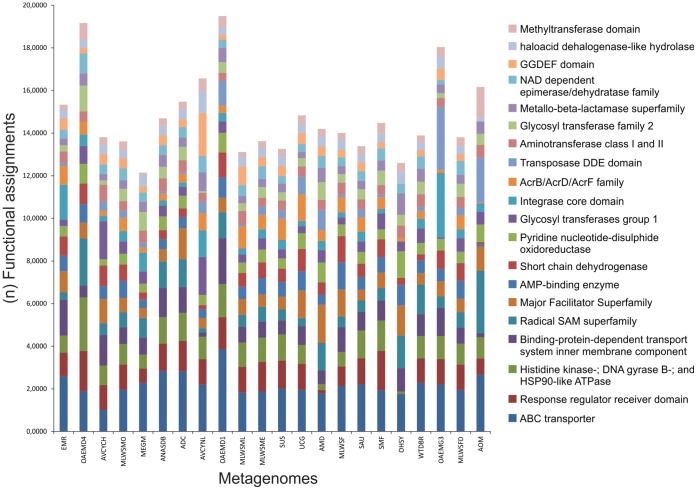
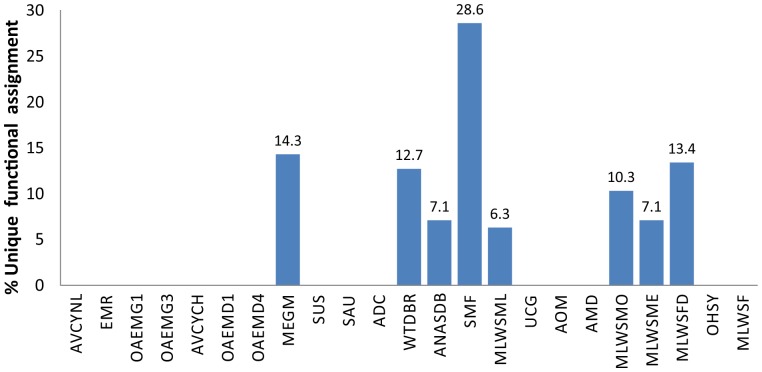
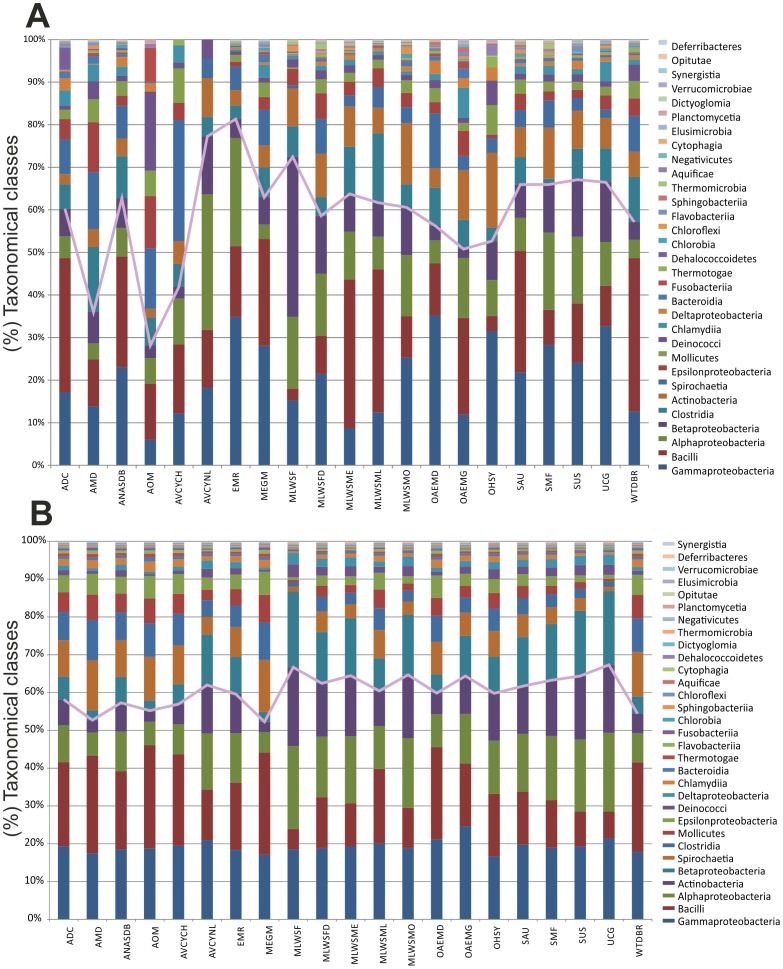
Environment-dependent genomic features have been defined for different metagenomes, whose genes and their associated processes are related to specific environments. Identification of ORFs and their functional categories are the most common methods for association between functional and environmental features. However, this analysis based on finding ORFs misses noncoding sequences and, therefore, some metagenome regulatory or structural information could be discarded. In this work we analyzed 23 whole metagenomes, including coding and noncoding sequences using the following sequence patterns: (G+C) content, Codon Usage (Cd), Trinucleotide Usage (Tn), and functional assignments for ORF prediction. Herein, we present evidence of a high proportion of noncoding sequences discarded in common similarity-based methods in metagenomics, and the kind of relevant information present in those. We found a high density of trinucleotide repeat sequences (TRS) in noncoding sequences, with a regulatory and adaptive function for metagenome communities. We present associations between trinucleotide values and gene function, where metagenome clustering correlate with microorganism adaptations and kinds of metagenomes. We propose here that noncoding sequences have relevant information to describe metagenomes that could be considered in a whole metagenome analysis in order to improve their organization, classification protocols, and their relation with the environment.
已经为不同的宏基因组定义了依赖环境的基因组特征,其基因及其相关过程与特定环境有关。识别 ORF 及其功能类别是将功能和环境特征联系起来的最常见方法。然而,这种基于寻找 ORF 的分析方法会忽略非编码序列,因此一些宏基因组的调控或结构信息可能会被丢弃。在这项工作中,我们使用以下序列模式分析了 23 个完整的宏基因组,包括编码和非编码序列:(G+C)含量、密码子使用 (Cd)、三核苷酸使用 (Tn) 和 ORF 预测的功能分配。在这里,我们证明了在宏基因组学中常用的基于相似性的方法中丢弃了大量非编码序列,以及这些序列中存在的相关信息。我们发现非编码序列中存在高密度的三核苷酸重复序列 (TRS),它们具有调控和适应宏基因组群落的功能。我们还发现了三核苷酸值与基因功能之间的关联,其中宏基因组聚类与微生物的适应和宏基因组的种类相关。我们在这里提出,非编码序列具有描述宏基因组的相关信息,可以在全宏基因组分析中考虑这些信息,以改善它们的组织、分类协议及其与环境的关系。