Institute of Technology, Tartu University, Nooruse 1, Tartu 50411, Estonia.
BMC Genomics. 2013 Apr 1;14:211. doi: 10.1186/1471-2164-14-211.
De novo genome sequencing of previously uncharacterized microorganisms has the potential to open up new frontiers in microbial genomics by providing insight into both functional capabilities and biodiversity. Until recently, Roche 454 pyrosequencing was the NGS method of choice for de novo assembly because it generates hundreds of thousands of long reads (<450 bps), which are presumed to aid in the analysis of uncharacterized genomes. The array of tools for processing NGS data are increasingly free and open source and are often adopted for both their high quality and role in promoting academic freedom.
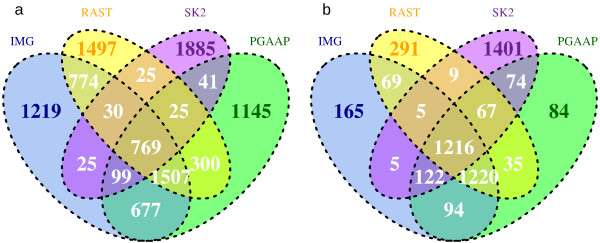
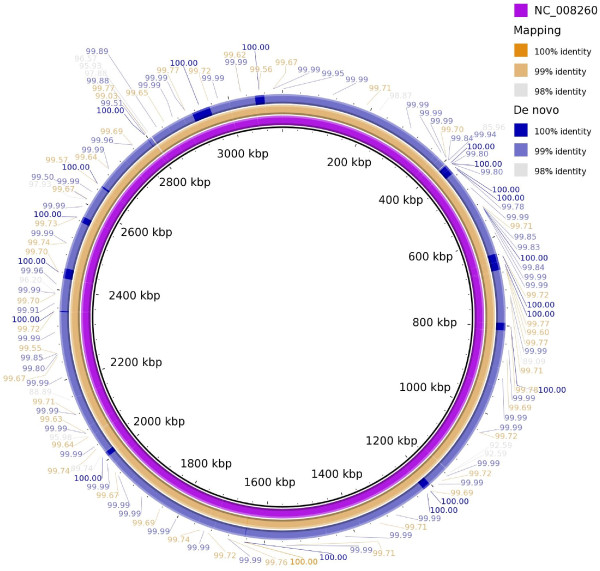
The error rate of pyrosequencing the Alcanivorax borkumensis genome was such that thousands of insertions and deletions were artificially introduced into the finished genome. Despite a high coverage (~30 fold), it did not allow the reference genome to be fully mapped. Reads from regions with errors had low quality, low coverage, or were missing. The main defect of the reference mapping was the introduction of artificial indels into contigs through lower than 100% consensus and distracting gene calling due to artificial stop codons. No assembler was able to perform de novo assembly comparable to reference mapping. Automated annotation tools performed similarly on reference mapped and de novo draft genomes, and annotated most CDSs in the de novo assembled draft genomes.
Free and open source software (FOSS) tools for assembly and annotation of NGS data are being developed rapidly to provide accurate results with less computational effort. Usability is not high priority and these tools currently do not allow the data to be processed without manual intervention. Despite this, genome assemblers now readily assemble medium short reads into long contigs (>97-98% genome coverage). A notable gap in pyrosequencing technology is the quality of base pair calling and conflicting base pairs between single reads at the same nucleotide position. Regardless, using draft whole genomes that are not finished and remain fragmented into tens of contigs allows one to characterize unknown bacteria with modest effort.
对以前未被描述的微生物进行从头基因组测序,有可能通过深入了解功能能力和生物多样性,为微生物基因组学开辟新的前沿。直到最近,罗氏 454 焦磷酸测序仍是从头组装的首选 NGS 方法,因为它生成了数十万条长读长(<450 bp),这些读长被认为有助于分析未被描述的基因组。用于处理 NGS 数据的工具套件越来越多是免费和开源的,并且经常因其高质量和在促进学术自由方面的作用而被采用。
对 Alcanivorax borkumensis 基因组进行焦磷酸测序的错误率导致数千个插入和缺失被人为地引入到完成的基因组中。尽管覆盖率很高(~30 倍),但它并没有允许参考基因组完全被映射。来自有错误的区域的reads 质量低、覆盖度低或缺失。参考映射的主要缺陷是通过低于 100%的一致性将人为的 indels 引入到 contigs 中,并由于人为的终止密码子而导致基因调用分散。没有组装程序能够执行与参考映射相当的从头组装。自动化注释工具在参考映射和从头草案基因组上的表现相似,并注释了从头组装的草案基因组中大多数 CDS。
用于 NGS 数据组装和注释的免费和开源软件(FOSS)工具正在迅速发展,以提供更少计算工作量的准确结果。可用性不是高优先级,这些工具目前不允许在没有人工干预的情况下处理数据。尽管如此,基因组组装程序现在可以轻松地将中等短读长组装成长 contigs(>97-98%的基因组覆盖率)。焦磷酸测序技术的一个显著缺陷是碱基对调用的质量以及在同一核苷酸位置处单读长之间的冲突碱基对。尽管如此,使用未完成且仍然碎片化为数十个 contigs 的草稿全基因组仍然可以让人们以适度的努力来描述未知细菌。