Medical Sciences, Indiana University School of Medicine, 1001 East 3rd St., Bloomington, IN 47405, United States.
Methods. 2013 Sep 15;63(2):126-34. doi: 10.1016/j.ymeth.2013.03.023. Epub 2013 Apr 1.
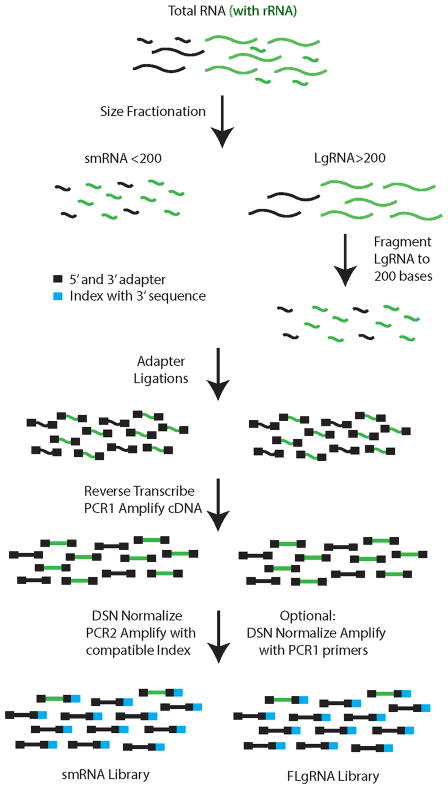
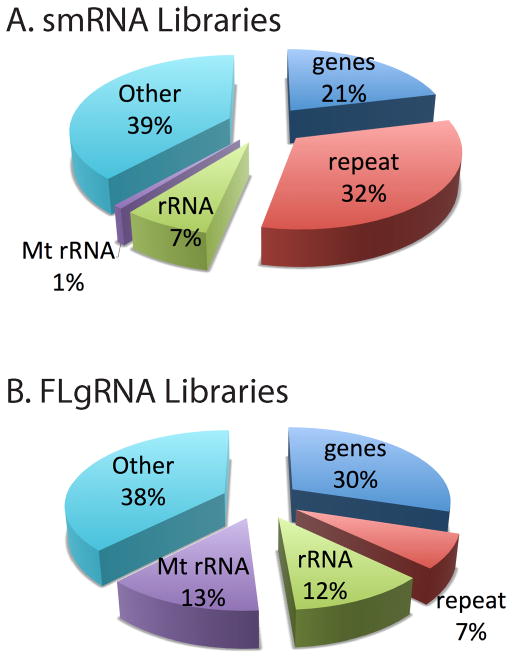
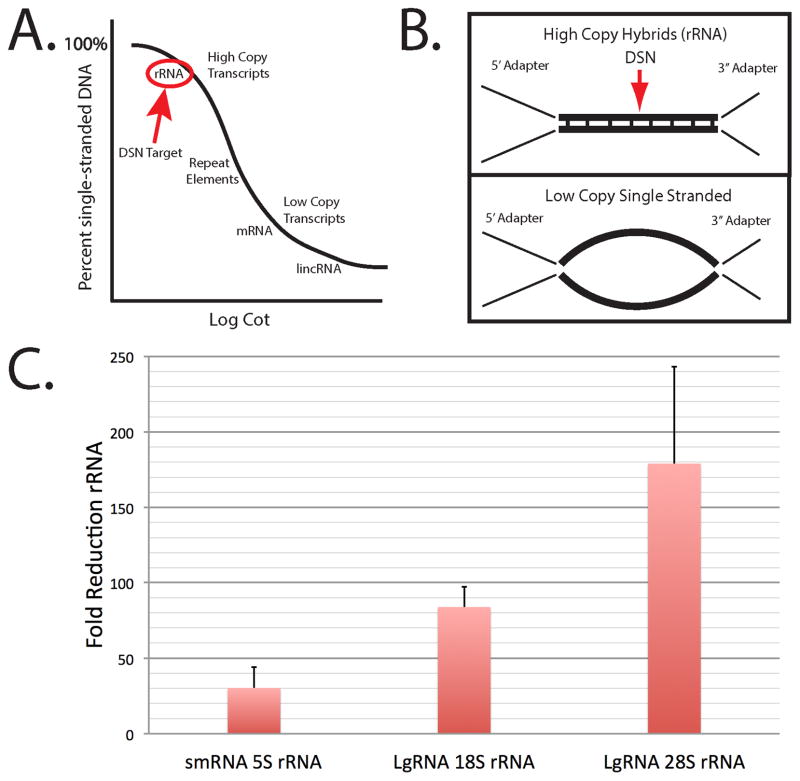
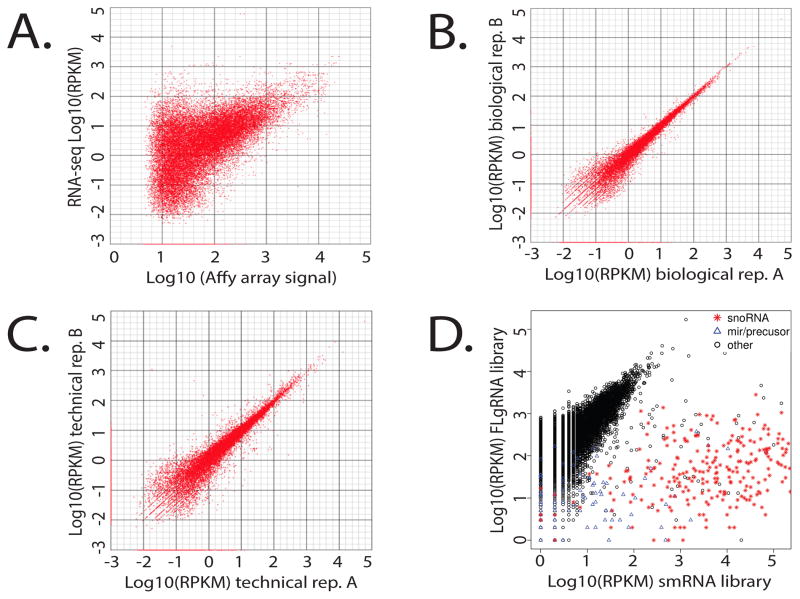
This report describes an improved protocol to generate stranded, barcoded RNA-seq libraries to capture the whole transcriptome. By optimizing the use of duplex specific nuclease (DSN) to remove ribosomal RNA reads from stranded barcoded libraries, we demonstrate improved efficiency of multiplexed next generation sequencing (NGS). This approach detects expression profiles of all RNA types, including miRNA (microRNA), piRNA (Piwi-interacting RNA), snoRNA (small nucleolar RNA), lincRNA (long non-coding RNA), mtRNA (mitochondrial RNA) and mRNA (messenger RNA) without the use of gel electrophoresis. The improved protocol generates high quality data that can be used to identify differential expression in known and novel coding and non-coding transcripts, splice variants, mitochondrial genes and SNPs (single nucleotide polymorphisms).
本报告描述了一种改进的方案,用于生成带有衔接子和条形码的 RNA-seq 文库,以捕获整个转录组。通过优化使用双链特异性核酸酶(DSN)从带有衔接子的条形码文库中去除核糖体 RNA 读段,我们证明了多路复用下一代测序(NGS)的效率得到了提高。这种方法可以检测所有 RNA 类型的表达谱,包括 miRNA(microRNA)、piRNA(Piwi 相互作用 RNA)、snoRNA(小核仁 RNA)、lincRNA(长非编码 RNA)、mtRNA(线粒体 RNA)和 mRNA(信使 RNA),而无需使用凝胶电泳。改进后的方案可生成高质量的数据,可用于识别已知和新的编码和非编码转录物、剪接变体、线粒体基因和单核苷酸多态性(SNP)的差异表达。