Persistent LABS, Persistent Systems Ltd., Pune, Maharashtra, India.
PLoS One. 2013 Apr 12;8(4):e60204. doi: 10.1371/journal.pone.0060204. Print 2013.
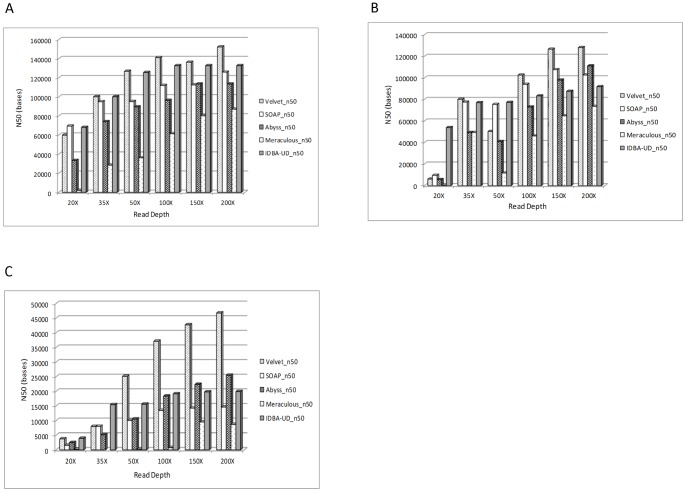
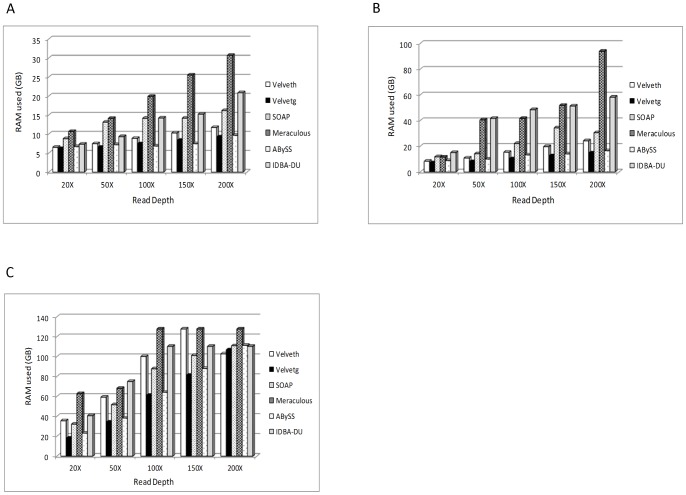
Next Generation Sequencing (NGS) is a disruptive technology that has found widespread acceptance in the life sciences research community. The high throughput and low cost of sequencing has encouraged researchers to undertake ambitious genomic projects, especially in de novo genome sequencing. Currently, NGS systems generate sequence data as short reads and de novo genome assembly using these short reads is computationally very intensive. Due to lower cost of sequencing and higher throughput, NGS systems now provide the ability to sequence genomes at high depth. However, currently no report is available highlighting the impact of high sequence depth on genome assembly using real data sets and multiple assembly algorithms. Recently, some studies have evaluated the impact of sequence coverage, error rate and average read length on genome assembly using multiple assembly algorithms, however, these evaluations were performed using simulated datasets. One limitation of using simulated datasets is that variables such as error rates, read length and coverage which are known to impact genome assembly are carefully controlled. Hence, this study was undertaken to identify the minimum depth of sequencing required for de novo assembly for different sized genomes using graph based assembly algorithms and real datasets. Illumina reads for E.coli (4.6 MB) S.kudriavzevii (11.18 MB) and C.elegans (100 MB) were assembled using SOAPdenovo, Velvet, ABySS, Meraculous and IDBA-UD. Our analysis shows that 50X is the optimum read depth for assembling these genomes using all assemblers except Meraculous which requires 100X read depth. Moreover, our analysis shows that de novo assembly from 50X read data requires only 6-40 GB RAM depending on the genome size and assembly algorithm used. We believe that this information can be extremely valuable for researchers in designing experiments and multiplexing which will enable optimum utilization of sequencing as well as analysis resources.
下一代测序(NGS)是一种颠覆性技术,在生命科学研究界得到了广泛认可。测序的高通量和低成本促使研究人员开展雄心勃勃的基因组项目,尤其是从头测序。目前,NGS 系统生成短读长序列数据,使用这些短读长进行从头基因组组装在计算上非常密集。由于测序成本降低和通量提高,NGS 系统现在能够以高深度对基因组进行测序。然而,目前尚无报告强调使用真实数据集和多种组装算法对高测序深度对基因组组装的影响。最近,一些研究评估了使用多种组装算法时序列覆盖率、错误率和平均读长对基因组组装的影响,但是,这些评估是使用模拟数据集进行的。使用模拟数据集的一个限制是,已知会影响基因组组装的变量,如错误率、读长和覆盖度,都被精心控制。因此,本研究旨在使用基于图的组装算法和真实数据集,确定不同大小基因组进行从头组装所需的最小测序深度。使用 SOAPdenovo、Velvet、ABySS、Meraculous 和 IDBA-UD 对大肠杆菌(4.6MB)、S.kudriavzevii(11.18MB)和 C.elegans(100MB)的 Illumina 读长进行了组装。我们的分析表明,除了需要 100X 读长深度的 Meraculous 之外,所有组装器都需要 50X 的最佳读深来组装这些基因组。此外,我们的分析表明,从头组装 50X 读长数据仅需要 6-40GB 的 RAM,具体取决于基因组大小和使用的组装算法。我们相信,对于设计实验和多路复用的研究人员来说,这些信息非常有价值,这将使测序以及分析资源得到最佳利用。