Department of Medicine, Case Western Reserve University, Cleveland, Ohio, United States of America. .
PLoS One. 2013 Apr 16;8(4):e62081. doi: 10.1371/journal.pone.0062081. Print 2013.
Pharmacologic inhibition of aldose reductase (AR) previously has been studied with respect to diabetic retinopathy with mixed results. Since drugs can have off-target effects, we studied the effects of AR deletion on the development and molecular abnormalities that contribute to diabetic retinopathy. Since recent data suggests an important role for leukocytes in the development of the retinopathy, we determined also if AR in leukocytes contributes to leukocyte-mediated death of retinal endothelial cells in diabetes.
Wild-type (WT; C57BL/6J) and AR deficient (AR(-/-)) mice were made diabetic with streptozotocin. Mice were sacrificed at 2 and 10 months of diabetes to evaluate retinal vascular histopathology, to quantify retinal superoxide production and biochemical and physiological abnormalities in the retina, and to assess the number of retinal endothelial cells killed by blood leukocytes in a co-culture system.
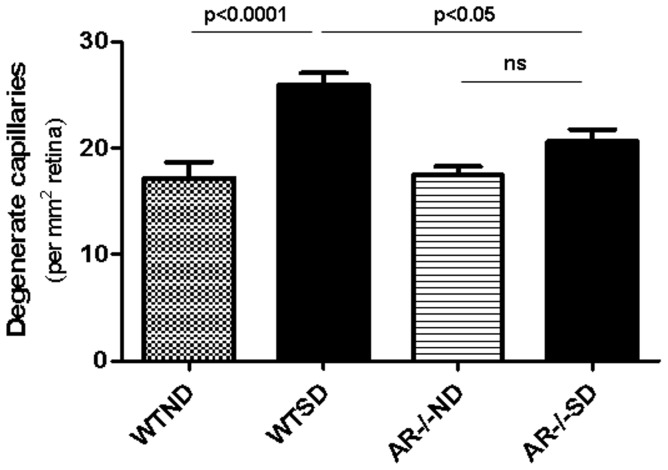
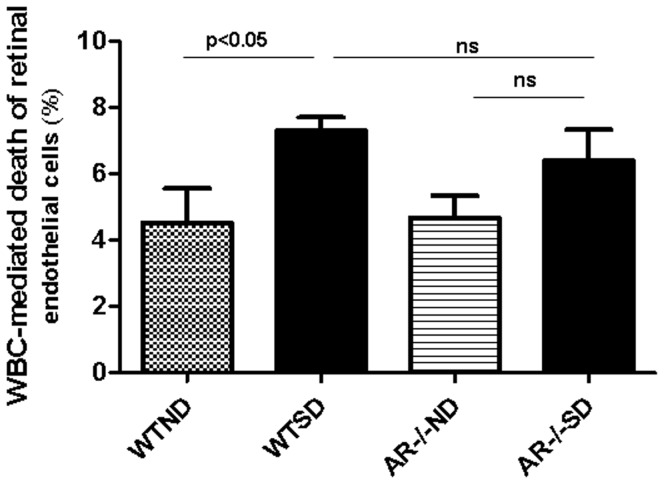
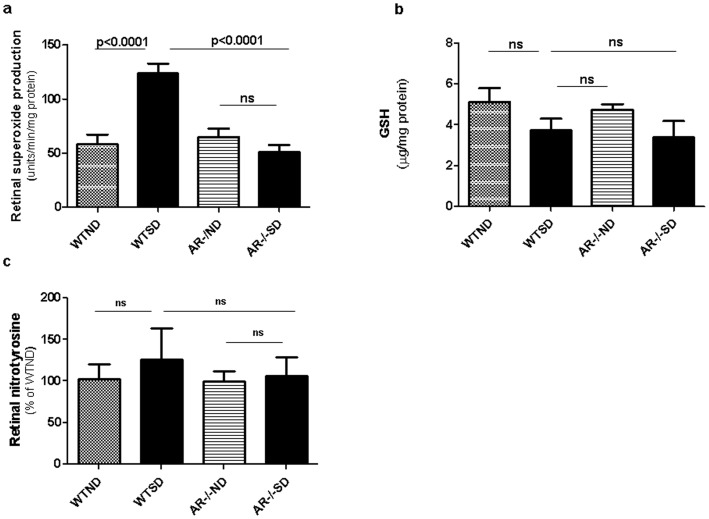
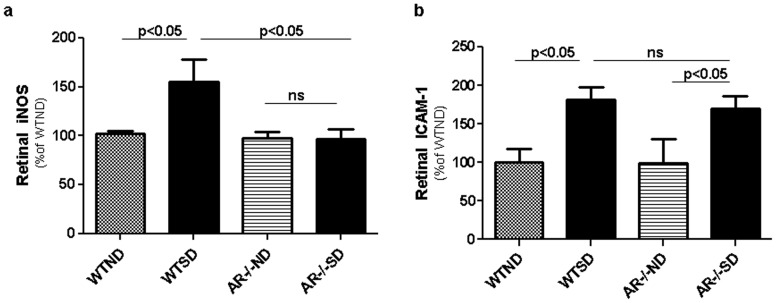
Diabetes in WT mice developed the expected degeneration of retinal capillaries, and increased generation of superoxide by the retina. Leukocytes from diabetic WT mice also killed more retinal endothelial cells than did leukocytes from nondiabetic animals (p<0.0001). Deletion of AR largely (P<0.05) inhibited the diabetes-induced degeneration of retinal capillaries, as well as the increase in superoxide production by retina. AR-deficiency significantly inhibited the diabetes-induced increase in expression of inducible nitric oxide synthase (iNOS) in retina, but had no significant effect on expression of intercellular adhesion molecule-1 (ICAM-1), phosphorylated p38 MAPK, or killing of retinal endothelial cells by leukocytes.
AR contributes to the degeneration of retinal capillaries in diabetic mice. Deletion of the enzyme inhibits the diabetes-induced increase in expression of iNOS and of superoxide production, but does not correct a variety of other pro-inflammatory abnormalities associated with the development of diabetic retinopathy.
醛糖还原酶(AR)的药理学抑制作用先前已在糖尿病性视网膜病变方面进行了研究,但结果不一。由于药物可能具有非靶向作用,因此我们研究了 AR 缺失对糖尿病性视网膜病变发展和导致其发生的分子异常的影响。由于最近的数据表明白细胞在视网膜病变的发生中起重要作用,我们还确定白细胞中的 AR 是否会导致糖尿病中白细胞介导的视网膜内皮细胞死亡。
使用链脲佐菌素使野生型(WT;C57BL/6J)和 AR 缺失(AR(-/-))小鼠发生糖尿病。在糖尿病 2 个月和 10 个月时处死小鼠,以评估视网膜血管组织病理学,定量视网膜中超氧阴离子的产生以及视网膜中的生化和生理异常,并评估在共培养系统中由血液白细胞杀死的视网膜内皮细胞的数量。
WT 小鼠的糖尿病导致视网膜毛细血管出现预期的退化,并增加了视网膜中超氧阴离子的产生。来自糖尿病 WT 小鼠的白细胞比来自非糖尿病动物的白细胞杀死更多的视网膜内皮细胞(p<0.0001)。AR 的缺失在很大程度上(P<0.05)抑制了糖尿病引起的视网膜毛细血管退化以及视网膜中超氧阴离子产生的增加。AR 缺失显著抑制了糖尿病诱导的视网膜中诱导型一氧化氮合酶(iNOS)表达的增加,但对细胞间黏附分子-1(ICAM-1)、磷酸化 p38MAPK 的表达或白细胞对视网膜内皮细胞的杀伤作用没有明显影响。
AR 促进了糖尿病小鼠视网膜毛细血管的退化。该酶的缺失抑制了糖尿病诱导的 iNOS 表达和超氧阴离子产生的增加,但不能纠正与糖尿病性视网膜病变发展相关的多种其他促炎异常。