Department of Plant Protection, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, PR China.
PLoS One. 2013 May 9;8(5):e61820. doi: 10.1371/journal.pone.0061820. Print 2013.
The sweetpotato whitefly, Bemisia tabaci (Hemiptera: Aleyrodidae), is one of the most widely distributed agricultural pests. Although it has developed resistance to many registered insecticides including the neonicotinoid insecticide thiamethoxam, the mechanisms that regulate the resistance are poorly understood. To understand the molecular basis of thiamethoxam resistance, "omics" analyses were carried out to examine differences between resistant and susceptible B. tabaci at both transcriptional and translational levels.
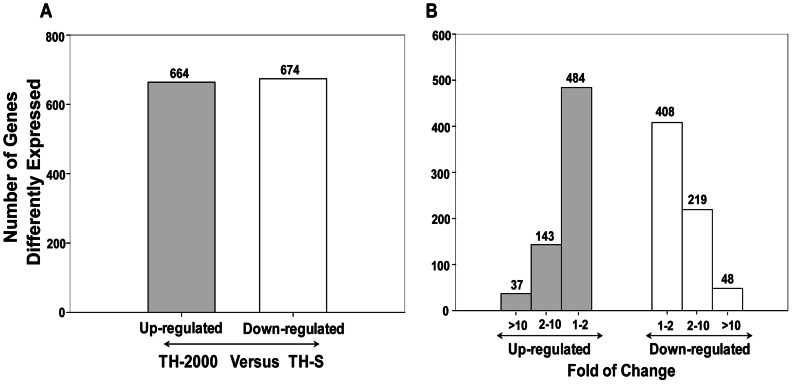
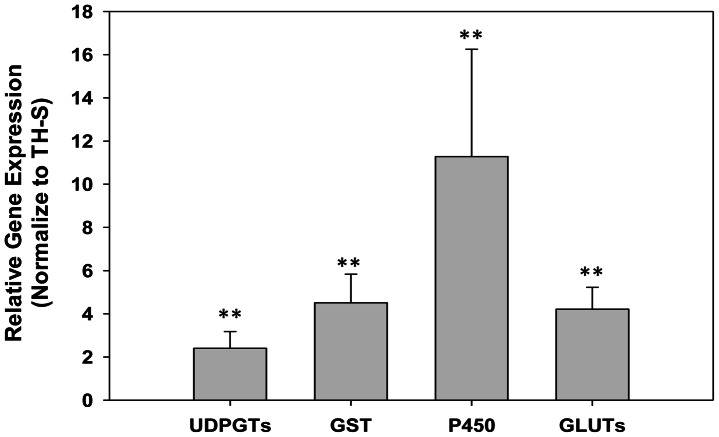
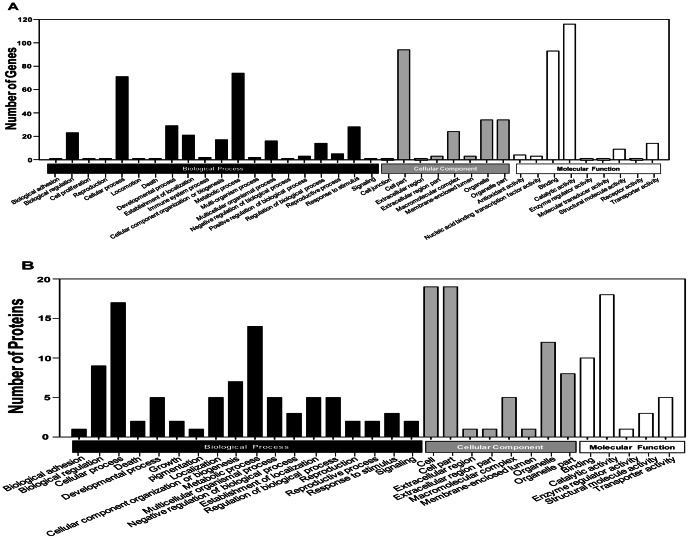
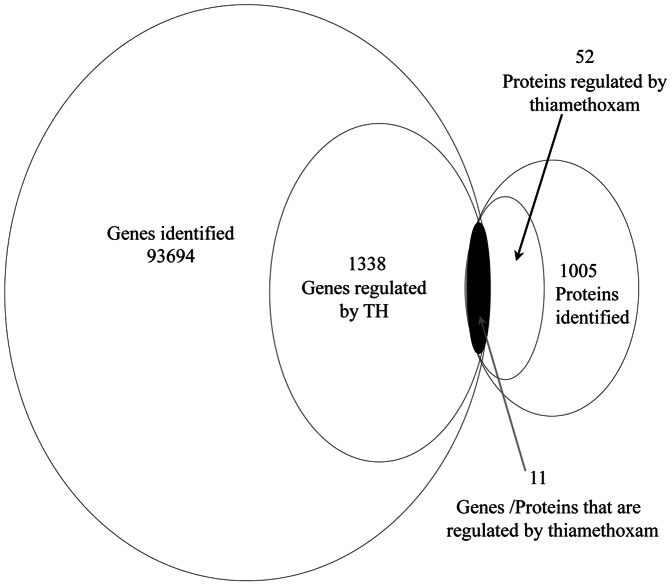
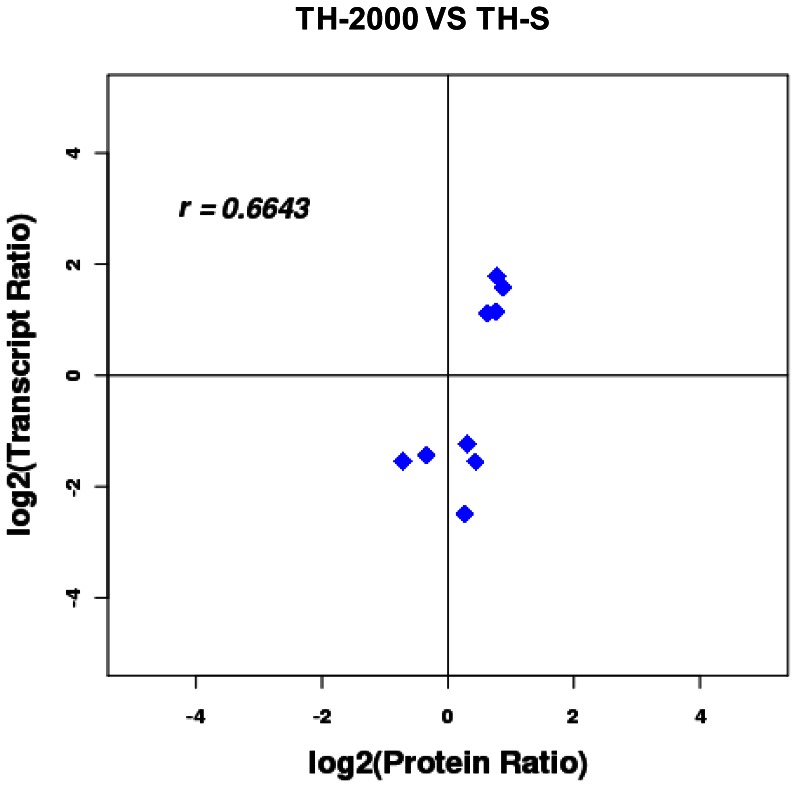
A total of 1,338 mRNAs and 52 proteins were differentially expressed between resistant and susceptible B. tabaci. Among them, 11 transcripts had concurrent transcription and translation profiles. KEGG analysis mapped 318 and 35 differentially expressed genes and proteins, respectively, to 160 and 59 pathways (p<0.05). Thiamethoxam treatment activated metabolic pathways (e.g., drug metabolism), in which 118 transcripts were putatively linked to insecticide resistance, including up-regulated glutathione-S-transferase, UDP glucuronosyltransferase, glucosyl/glucuronosyl transferase, and cytochrome P450. Gene Ontology analysis placed these genes and proteins into protein complex, metabolic process, cellular process, signaling, and response to stimulus categories. Quantitative real-time PCR analysis validated "omics" response, and suggested a highly overexpressed P450, CYP6CX1, as a candidate molecular basis for the mechanistic study of thiamethoxam resistance in whiteflies. Finally, enzymatic activity assays showed elevated detoxification activities in the resistant B. tabaci.
This study demonstrates the applicability of high-throughput omics tools for identifying molecular candidates related to thiamethoxam resistance in an agricultural important insect pest. In addition, transcriptomic and proteomic analyses provide a solid foundation for future functional investigations into the complex molecular mechanisms governing the neonicotinoid resistance in whiteflies.
甘薯粉虱,烟粉虱(半翅目:粉虱科),是分布最广泛的农业害虫之一。尽管它已经对许多已注册的杀虫剂产生了抗性,包括新烟碱类杀虫剂噻虫嗪,但调节抗性的机制仍知之甚少。为了了解噻虫嗪抗性的分子基础,我们进行了“组学”分析,以检查抗性和敏感烟粉虱在转录和翻译水平上的差异。
在抗性和敏感烟粉虱之间,共有 1338 个 mRNA 和 52 个蛋白差异表达。其中,有 11 个转录本具有同时的转录和翻译谱。KEGG 分析将 318 个和 35 个差异表达的基因和蛋白质分别映射到 160 个和 59 个途径(p<0.05)。噻虫嗪处理激活了代谢途径(例如,药物代谢),其中 118 个转录本被认为与杀虫剂抗性有关,包括上调的谷胱甘肽-S-转移酶、UDP 葡萄糖醛酸基转移酶、葡萄糖基/葡萄糖醛酸基转移酶和细胞色素 P450。基因本体论分析将这些基因和蛋白质归入蛋白质复合物、代谢过程、细胞过程、信号和对刺激的反应类别。定量实时 PCR 分析验证了“组学”反应,并表明一种高度过表达的 P450,CYP6CX1,是噻虫嗪抗性机制研究的候选分子基础。最后,酶活性测定显示抗性烟粉虱的解毒活性升高。
本研究证明了高通量组学工具在鉴定与农业重要害虫噻虫嗪抗性相关的分子候选物方面的适用性。此外,转录组学和蛋白质组学分析为未来研究新烟碱类杀虫剂抗性的复杂分子机制提供了坚实的基础。